CONTENTS
1. Hyperemia &Congestion
2. Edema
Increased Hydrostatic Pressure Reduced Plasma Osmotic Pressure
Lymphatic Obstruction Sodium & Water Retention Inflammation
3. Hemorrhage Hemostasis: Normal Hemostasis
4. Thrombosis Disseminated Intravascular Coagulation (DIC)
5. Embolism Pulmonary & Systemic Thromboembolism
Fat Embolism Amniotic Fluid Embolism Air Embolism
6. Infarction: Factors That Influence Infarct Development
7. Shock: Stages of Shock
8. 10. Pathogenesis of Septic Shock
3.
LEARNING OBJECTIVES
At theend of this lecture series, the students should be able to define the
following terms:
Hyperaemia Congestion Edema Haemorrhage Hemostasis
Thrombosis Embolism Infarction Shock
Understand the pathophysiologic categories, morphology & the clinical profile
of edema
Be able to differentiate between exudate & transudate
Be able to define hemostasis & the sequence of events in normal hemostasis
Define thrombosis, its causes, morphology & the fate of a thrombus
Be able to differentiate between arterial & venous thrombi
4.
• Be ableto differentiate between Antemortem thrombi & postmortem clot
• Be able to define an embolus, its causes & types
• Understand the causes & morphology of both pulmonary & systemic
thromboembolism
• Be able to define infarction, its causes, classification & the factors that
influence its development
• Be able to define & classify shock , its causes & the their mechanisms
• Be able to in detail describe the pathophysiology of septic shock
• Understand the different stages of shock & its associated morphology
5.
INTRODUCTION
• The healthof cells and tissues depends on the circulation of blood which delivers oxygen &
nutrients & removes wastes generated by cellular metabolism.
• In normal conditions of blood passing through the capillary beds, proteins in the plasma are
retained within the vasculature & there is little net movement of water & electrolytes into
the tissues.
• This balance is often disturbed by pathologic conditions with the following consequences:
• Alteration of endothelial function
• Increase vascular hydrostatic pressure
• Decrease plasma protein content
• The result is the accumulation of fluid in tissues resulting from a net movement of water
into extravascular spaces called Edema
6.
Hemostasis: The processof blood clotting that prevents excessive
bleeding after blood-vessel damage. Inadequate hemostasis may result
in Hemorrhage which can compromise regional tissue perfusion
• If haemorrhage is massive & rapid, it may lead to hypotension, shock
& death.
• Inappropriate clotting leads to Thrombosis or Embolism (Migration of
clots)
• If blood vessels are obstructed, it can result in Infarction of tissues
(Ischemic cell death)
7.
HYPEREMIA & CONGESTION
Hyperemia
•An increase in blood volume within a tissue
• An active process resulting from arteriolar dilation & increased blood inflow e.g. Inflammation
• The tissues are redder than normal because of engorgement with oxygenated blood.
Congestion
• An increase in blood volume within a tissue
• Passive process resulting from impaired outflow of venous blood from a tissue.
• It can occur systemically (Congestive heart failure)
• It can occur locally from isolated venous obstruction
• Congested tissues show cyanosis (Abnormal blue-red color) due to accumulation of deoxygenated
hemoglobin
8.
MORPHOLOGY
Gross: Cut surfacesof hyperemic or congested tissues feel wet & typically ooze blood
Microscopic
1. In acute pulmonary congestion: Blood-engorged alveolar capillaries, alveolar septal edema & intra-alveolar
hemorrhage.
2. In chronic pulmonary congestion: Thickened & fibrotic alveolar septa & the alveolar spaces contain
numerous hemosiderin laden macrophages called “Heart failure cells” derived from phagocytosed red cells.
These pigments stain blue with the Prussian blue stain called Perl’s stain.
3. In acute hepatic congestion the central vein & sinusoids are distended with blood
4. In chronic passive congestion of the liver: The central regions of the hepatic lobules are red-brown & slightly
depressed (Cell loss) within surrounding zones of uncongested tan a times fatty, liver called Nutmeg liver.
• Right-sided heart failure is the most common cause.
Gross: Enlarged liver with a tense capsule.
Microscopic: Centrilobular hepatocyte necrosis, hemorrhage & hemosiderin-laden macrophages
10.
EDEMA
a. Defined asabnormal increase in interstitial fluid within tissues
b. 60% of lean body weight is water
c. Two thirds is intracellular
d. One third is in the extracellular compartments in the form of interstitial fluid
e. 5% of the body’s water is in blood plasma.
f. Extravascular fluid can collect in body cavities collectively called Effusions
g. Anasarca is severe, generalized edema marked by profound swelling of
subcutaneous tissues & accumulation of fluid in body cavities.
11.
The abnormal fluidcollects in different body cavities:
1. Pleural cavity (Hydrothorax)
2. Pericardial cavity (Hydropericardium)
3. Peritoneal cavity (Hydroperitoneum or ascites).
Edema can be due to:
a. Inflammatory largely related to increased vascular permeability
b. Non-inflammatory
Types of edema: Localized or generalized
Localized (Causes)
1. Inflammation: Mainly from increased permeability of the blood vessels
2. Immune reaction
3. Venous/Lymphatic obstruction: Venous thrombus in the leg veins/Filariasis, post-irradiation/post-surgical
or neoplastic.
12.
Generalized (Causes)
• Mainlyfrom disorders of fluid & electrolyte balance
• Anarsaca is a severe & generalized form of edema with subcutaneous tissue swelling & accumulation in
the visceral organs & body cavities.
Causes:
1. Heart failure: Most common cause of generalized edema
2. Renal disease: Associated with loss of serum proteins into the urine like in Nephrotic syndrome
3. Liver cirrhosis: From decreased synthesis of albumin
Fluid movement between the vascular & interstitial spaces is governed mainly by two opposing forces
1. Vascular hydrostatic pressure
2. Colloid osmotic pressure produced by plasma proteins
• Normally outflow of fluid by hydrostatic pressure at the arteriolar end of the microcirculation is nearly
balanced by inflow at the venular end owing to elevated osmotic pressure
13.
1. Thus thereis only a small net outflow of fluid into the interstitial space which is
drained by lymphatic vessels.
2. Either increased hydrostatic pressure or diminished colloid osmotic pressure
causes increased movement of water into the interstitium
3. The edema fluid of increased hydrostatic pressure or reduced intravascular
colloid typically is a protein-poor transudate
4. The edema fluid from increased vascular permeability (Inflammatory) is a
protein-rich exudate with a high specific gravity.
5. Transudate: Defined as a protein poor fluid. (Edema caused by increased
hydrostatic pressure). Examples are seen in Heart, Renal or Liver failure.
6. Exudate: Defined as a protein rich fluid. (Edema of inflammation)
PATHOPHYSIOLOGIC CATEGORIES OFEDEMA
1. Increased capillary hydrostatic pressure(HP): Increases in HP are mainly caused by disorders that impair venous
return
• Impaired venous return like congestive heart failure (CHF)
• Ascites from liver cirrhosis
• Venous obstruction/Compression like thrombosis, external pressures, constrictive pericarditis, deep venous
thrombosis in the lower extremity
• Secondary hyperaldosteronism is a common feature of generalized edema
2. Reduced plasma osmotic or oncotic pressure (Hypo-proteinemia): Reduction of plasma albumin concentrations
leads to decreased colloid osmotic pressure of the blood & loss of fluid from the circulation
• Malnutrition
• Nephrotic syndrome: Most important cause of albumin loss from the blood.
• Liver cirrhosis: Reduced albumin synthesis
• Note that low albumin levels lead to edema, reduced intravascular volume, renal hypoperfusion & secondary
hyperaldosteronism
16.
3. Lymphatic obstruction:Compromises resorption of fluid from interstitial spaces.
• Inflammatory (Filariasis): Massive edema of the lower extremity & external genitalia called “elephantiasis” by
producing inguinal lymphatic & lymph node fibrosis
• Post irradiation/surgical
• Neoplastic
• External pressure
• Hereditary (Milroy's disease)
4. Sodium & water retention
• Excessive salt intake with renal insufficiency
• Increased salt intake
• Abnormal secretion of aldosterone
• Post-streptococcal glomerulonephritis
• Acute renal failure
5. Inflammation: Acute inflammation & chronic inflammation
17.
MORPHOLOGY OF EDEMA
Edemamost commonly is encountered in subcutaneous tissues, lungs & brain.
1. Subcutaneous edema: Typically most pronounced in the legs (Standing) & sacrum
(Recumbence) called dependent edema.
• Finger pressure over edematous subcutaneous tissue displaces the interstitial fluid, leaving a
finger-shaped depression called pitting edema.
• Edema resulting from renal dysfunction or Nephrotic syndrome manifests first in loose
connective tissues especially the eyelids (Periorbital edema)
2. Pulmonary edema: Increased weight of the lungs X2-3 normal weight cut section shows
frothy, a times blood-tinged fluid consisting of a mixture of air, edema fluid & extravasated red
cells
3. Brain edema: Localized (Abscess or tumour) or generalized. With generalized (Narrowed sulci
& swollen & flattened gyri swell (Against the skull).
18.
Clinical profile ofedema
1. Cardiac edema: Usually from CHF
• Causes dependent pitting edema mostly in lower extremities in ambulatory & sacral in bed ridden
patients.
• Fluid also occurs in the serous cavities
2. Renal edema: Nephrotic syndrome, acute tubular injury, acute glomerulonephritis.
• More severe & marked in the Peri-orbital tissues (Least resistance) ankles & genitalia
3. Pulmonary edema
• Most important form of localized edema from elevated pulmonary hydrostatic pressure & increased
permeability of the alveolar capillaries
• Seen with Lt sided cardiac failure, mitral stenosis, high altitudes, acute respiratory distress syndrome,
fulminant infections damaging the alveolar capillaries
19.
• Edema isconfined to the basal regions of the lungs & X-rays show linear lines called Kelly B-
lines which indicates dilated lymphatic's
• d. The lungs are heavy, moist, sub-crepitant, with pinkish frothy fluid on pressing a slice of
the lungs
• e. Microscopically the alveoli are filled with proteinaceous edema fluid with congestion
4. Cerebral edema
• Life threatening as the brain is encased in a bony skull cage
• There are 4 types: Vasogenic, Cytotoxic or cellular, osmotic & interstitial
• Vasogenic: (An accumulation of fluid on the outside of the brain cells due to disturbances of
the blood brain barrier) is the commonest & can be seen with contusion & infarction of the
brain, brain tumours & abscesses
•
20.
Clinical Features ofedema
• The effects of edema vary from asymptomatic to rapidly fatal.
• Subcutaneous edema signals potential underlying cardiac or renal disease
• It also can impair wound healing and the clearance of infections.
• Pulmonary edema is seen most frequently with left ventricular failure,
renal failure, and acute respiratory distress syndrome, inflammatory &
infectious disorders of the lung.
• It can cause death via raised intracranial pressure by interfering with brain
stem vascular supply (Compressed) & injury to the medullary centers
controlling respiration & other vital functions
21.
HEMORRHAGE
Defined as extravasationof blood from vessels most often the result of damage to blood vessels or defective
clot formation.
Other causes
• Capillary bleeding in chronically congested tissues
• Trauma
• Atherosclerosis
• Inflammatory or neoplastic erosion of a vessel wall
• Hemorrhagic diatheses: Inherited or acquired defects in vessel walls, platelets or coagulation factors
• Accumulate within a tissue as a hematoma as a bruise or fatal massive retroperitoneal hematoma (Rupture
dissecting aortic aneurysm)
• Large bleeds into body cavities: Hemothorax, Hemopericardium, Hemoperitoneum, or Hemarthrosis (Joints).
• Extensive hemorrhages can result in jaundice from massive breakdown of red cells & hemoglobin
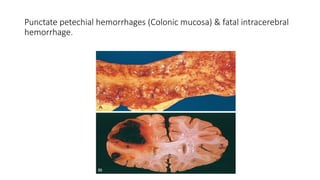
Petechiae: Minute 1-2mm in diameter hemorrhages into skin, mucous membranes or serosal surfaces
Causes
• low platelet counts (thrombocytopenia)
• defective platelet function
• loss of vascular wall support, as in vitamin C deficiency
Purpura: 3-5 mm hemorrhages.
Causes
• Same as above
• Trauma
• vascular inflammation (Vasculitis)
• Increased vascular fragility
• Ecchymoses: 1-2 cm subcutaneous hematomas (Called bruises).
24.
Clinical significance
• Dependson the volume of blood lost
• Rate of bleeding.
• Hemorrhagic (Hypovolemic) shock
• The site of hemorrhage also is important e.g. between subcutaneous
tissues or the brain
• Iron deficiency anemia from chronic or recurrent external blood loss
Note that iron is efficiently recycled from phagocytosed red cells thus
internal bleeding like a hematoma does not lead to iron deficiency anaemia
25.
HEMOSTASIS
• Normal hemostasiscomprises a series of regulated processes that culminate in the formation of a
blood clot that limits bleeding from an injured vessel.
• The pathologic counterpart of hemostasis is thrombosis: Formation of blood clot (Thrombus) within
non-traumatized, intact vessels.
NORMAL HEMOSTASIS
Hemostasis is a process involving platelets, clotting factors & endothelium that occurs at the site of
vascular injury leading to the formation of a blood clot to prevent or limit the extent of bleeding.
• The general sequence of events leading to hemostasis at a site of vascular injury is as follows:
1. Arteriolar vasoconstriction: Occurs immediately & markedly reduces blood flow to the injured area,
mediated by reflex neurogenic mechanisms & augmented by the local secretion of factors such as
endothelin, a potent endothelium-derived vasoconstrictor.
This effect is transient because bleeding would resume if platelets are not activated & coagulation factors
are not brought into play.
26.
2. Primary hemostasis:Formation of the platelet plug. Disruption of the endothelium
exposes subendothelial von Willebrand factor (vWF) & collagen, which promote platelet
adherence & activation.
Activation of platelets results in a dramatic shape change (From small rounded discs to flat
plates with spiky protrusions that markedly increased surface area), as well as the release
of secretory granules.
Within minutes the secreted products recruit additional platelets which undergo
aggregation to form a primary hemostatic plug
3. Secondary hemostasis: Deposition of fibrin. Vascular injury exposes Tissue factor at the
site of injury. Tissue factor is a membrane-bound procoagulant glycoprotein that is
normally expressed by subendothelial cells in the vessel wall such as smooth muscle cells
& fibroblasts. Tissue factor binds & activates factor VII, setting in motion a cascade of
reactions that leads to the generation of Thrombin.
27.
Thrombin cleaves circulatingfibrinogen into insoluble fibrin, creating a fibrin meshwork. It is also a
potent activator of platelets leading to additional platelet aggregation at the site of injury.
(Secondary hemostasis consolidates the initial platelet plug)
4. Clot stabilization & resorption: Polymerized fibrin & platelet aggregates undergo contraction to
form a solid, permanent plug that prevents further hemorrhage.
• At this stage, counter regulatory mechanisms (Tissue plasminogen activator (t-PA) made by
endothelial cells) are set into motion that limit clotting to the site of injury & eventually lead to
clot resorption & tissue repair.
SUMMARY
1. Vasoconstriction
2. Platelet activation & aggregation
3. Activation of clotting factors & formation of fibrin
4. Clot resorption
28.
SEQUENCE OF EVENTSIN NORMAL HEMOSTASIS
1. Injury to the vessel wall
2. Transient vasoconstriction
3. Endothelial injury
4. Exposure of thrombogenic subendothelium
5. Adherence of platelets to subendothelium
6. Activation of platelets
7. Release reaction of platelet & aggregation
8. Primary platelet plug
9. Release of tissue factors
10. Activation of the coagulation cascade
11. Formation of fibrin clot (Secondary hemostatic plug)
29.
ASSIGNMENT
What are theroles of platelets, coagulation factors & endothelium in
hemostasis?
•
30.
THROMBOSIS
The primary abnormalitiesthat lead to intravascular thrombosis are:
1. Endothelial injury
2. Stasis or turbulent blood flow
3. Hypercoagulability of the blood
These three form what is called the “Virchow Triad?”
a. Endothelial injury leading to platelet activation almost inevitably underlies thrombus formation
b. Platelet adherence & activation is a necessary prerequisite for thrombus formation
c. Severe endothelial injury may trigger thrombosis by exposing VWF & tissue factor
d. Inflammation & physical injury, infectious agents, abnormal blood flow, inflammatory mediators,
metabolic abnormalities (Hypercholesterolemia, Homocystinemia) & toxins promote thrombosis by
changing the pattern of gene expression in endothelium to one that is “Prothrombotic” called
endothelial activation or dysfunction
Prothrombotic alterations
1. Procoagulantchanges
a. Activated endothelial cells (cytokines) down regulate the expression of
Thrombomodulin (Modulator of thrombin activity) leading to sustained activation
of thrombin, which can in turn stimulate platelets & augment inflammation
b. Inflamed endothelium also down regulates the expression of other anticoagulants
(Protein C & tissue factor protein inhibitor promoting a procoagulant state
2. Anti-fibrinolytic effects
Activated endothelial cells secrete Plasminogen activator inhibitors (PAI), which
limit fibrinolysis & down regulate the expression of t-PA, favoring the development
of thrombi.
33.
ABNORMAL BLOOD FLOW
a.Turbulence causes endothelial injury or dysfunction, forms countercurrents & local pockets of
stasis.
b. Stasis is a major factor in the development of venous thrombi.
c. Note that in normal laminar blood flow, platelets (Other blood cells) are found mainly in the
center of the vessel lumen separated from the endothelium by a slower-moving layer of plasma.
Consequences of stasis & turbulence
a. Both promote endothelial cell activation & enhanced procoagulant activity in part through
flow-induced changes in endothelial gene expression
b. Stasis allows platelets & leukocytes to come into contact with the endothelium
c. Stasis slows the washout of activated clotting factors & impedes the inflow of clotting factor
inhibitors.
34.
HYPERCOAGULABILITY
Defined as anabnormally high tendency of the blood to clot typically caused by alterations in coagulation
factors.
a. It is an important underlying risk factor for venous thrombosis.
b. The alterations of the coagulation pathways can be divided into primary (Genetic) & secondary (Acquired)
disorders
c. Primary (Inherited) hypercoagulability is most often caused by mutations in the factor V & Prothrombin
genes
d. About 2-15% of Caucasians carry a specific factor V mutation called the Leiden mutation
e. The frequency of this mutation approaches 60% in those with recurrent deep venous thrombosis (DVT)
f. The mutation alters an amino acid residue in factor V rendering it resistant to proteolysis by protein C.
(Anti-thrombotic counter-regulatory mechanism)
g. Heterozygotes carry X5-fold increased risk for venous thrombosis while homozygotes carry X50-fold
increased risk
MORPHOLOGY
a. Thrombi candevelop anywhere in the cardiovascular system.
b. Arterial or cardiac thrombi typically arise at sites of endothelial injury or turbulence
c. Venous thrombi characteristically occur at sites of stasis.
d. Thrombi are focally attached to the underlying vascular surface & tend to propagate toward the heart
(Arterial thrombi grow in a retrograde direction from the point of attachment while venous thrombi
extend in the direction of blood flow).
e. The propagating portion of a thrombus tends to be poorly attached & thus prone to fragmentation &
embolization.
f. Thrombi can have gross & microscopically apparent laminations called lines of Zahn representing pale
platelet & fibrin layers alternating with darker red cell-rich layers. Significance: Are only found in thrombi
that form in flowing blood & it distinguish Antemortem thrombosis from the bland non-laminated clots
that form in the postmortem state.
g. Thrombi in heart chambers or the aortic lumen are called mural thrombi.
37.
Arterial thrombi
a. Arefrequently occlusive.
b. They are typically rich in platelets
c. Usually superimposed on a ruptured atherosclerotic plaque
Venous thrombi (Phlebothrombosis)
a. Are almost invariably occlusive
b. Frequently propagate some distance toward the heart forming long cast within the
vessel lumen that is prone to give rise to emboli (Contain more enmeshed red cells
called moniker red or stasis thrombi).
c. The veins of the lower extremities are most commonly affected (90% of venous
thromboses)
38.
a. Thrombi onheart valves are called vegetations.
b. Bacterial or fungal blood borne infections can cause valve damage leading to the
development of large thrombotic masses (Infective endocarditis)
c. Sterile vegetations can develop on non-infected valves in hypercoagulable states
(“Nonbacterial thrombotic endocarditis”
d. Less commonly, sterile, verrucous endocarditis (LibmanSacks endocarditis) can occur in
the setting of systemic lupus erythematosus
Chicken fat appearance: Is as a result of settling of the red cell at the bottom of the clot
(Dark red dependent bottom) with a superficial yellowish fatty material (“chicken fat”
upper portion)
39.
DIFFERENCES BETWEEN ANTEMORTEMTHROMBI AND POSTMORTEM CLOTS
ANTEMORTEM THROMBI POSTMORTEM CLOT
Granular Soft
Firm Gelatinous
Red Reddish brown in colour
Focally attached to the vessel wall Usually not attached to the underlying vessel wall
Presence of lines of Zahn Absent
No chicken fat appearance Chicken fat appearance present
Underlying infarcted areas Normal endocardium
Contain gray strands of deposited fibrin. Does not
40.
FATE OF THETHROMBUS
Evolves through 4 processes
1. Propagation: The thrombus enlarges from additional platelets & fibrin, increasing the odds of vascular occlusion or
embolization.
2. Embolization: Part or all of the thrombus is dislodged & transported e in the vasculature
3. Dissolution: If a thrombus is newly formed, activation of fibrinolytic factors may lead to its rapid shrinkage & complete
dissolution. With older thrombi, extensive fibrin polymerization renders the thrombus substantially more resistant to
plasmin-induced proteolysis, and lysis is ineffectual.
4. Organization & recanalization
• Older thrombi become organized by the ingrowth of endothelial cells, smooth muscle cells & fibroblasts.
• With time capillary channels are formed to re-establish the continuity of the original lumen.
• Further recanalization can sometimes convert a thrombus into avascularized mass of connective tissue that is eventually
incorporated into the wall of the remodeled vessel
• A times, instead of organizing, the center of a thrombus may undergo enzymatic digestion from the release of lysosomal
enzymes from entrapped leukocytes. With bacterial seeding, infection occurs which may weaken the vessel wall leading
to the formation of Mycotic aneurysm
41.
Venous Thrombosis (Phlebothrombosis)
a.Most occur in the superficial or the deep veins of the leg.
b. Superficial: Usually arise in the saphenous system especially in the setting of varicosities;
c. Rarely embolize but painful causing local congestion & swelling from impaired venous outflow,
predisposing the overlying skin to the development of infections & varicose ulcers.
Deep venous thromboses (DVTs)
• Occur in the larger leg veins at or above the knee joint (Popliteal, femoral & iliac)
• Prone to embolize.
• Causes local pain & edema
• Collateral channels often circumvent the venous obstruction.
• Usually DVTs are entirely asymptomatic in about 50% of cases
• Embolization is to the lungs.
• Lower-extremity DVTs are associated with stasis & hypercoagulable states
42.
Predisposing factors
• Congestiveheart failure
• Prolonged bed rest
• Immobilization
• Trauma, surgery & burns immobilize a patient & are associated with vascular injury, procoagulant
release, increased hepatic synthesis of coagulation factors & reduced t-PA production.
• Pregnancy: Amniotic fluid infusion into the circulation at delivery, pressure from enlarging fetus &
uterus produce stasis in the leg veins of the legs
• Late pregnancy & postpartum period are associated with hypercoagulability.
• Tumor associated procoagulant release in disseminated cancers called migratory
thrombophlebitis or Trousseau syndrome,
• DVT is increased in persons older than 50 years.
43.
Arterial & CardiacThrombosis
• Atherosclerosis is a major cause of arterial thromboses associated with loss of endothelial integrity &
abnormal blood flow
• Myocardial infarction can predispose to cardiac mural thrombi from dyskinetic myocardial contraction &
endocardial injury
• Rheumatic heart disease (Atrial mural thrombi) from atrial dilation & fibrillation.
• Cardiac & aortic mural thrombi are prone to embolization.
• Brain, kidneys & spleen are particularly prone due to their rich blood supply.
DISSEMINATED INTRAVASCULAR COAGULATION (DIC)
Defined as widespread thrombosis within the microcirculation
a. Seen in many disorders like obstetric complications to advanced malignancy.
b. The widespread microvascular thrombosis consumes platelets & coagulation proteins (Synonym consumptive
coagulopathy)
c. Fibrinolytic mechanisms are activated.
44.
EMBOLISM
An embolus isa detached intravascular solid, liquid, or gaseous mass carried by the blood from its
point of origin to a distant site (Causes tissue dysfunction or infarction).
• Most are derived from a dislodged thrombus (Thromboembolism).
Other types are:
• Fat droplets, Air or nitrogen bubbles, Atherosclerotic debris (Cholesterol emboli), Tumour
fragments, bone marrow fragments or amniotic fluid.
• Emboli lodge in vessels too small to permit further passage resulting in partial or complete
vascular occlusion
• Depending on the site of origin emboli can lodge anywhere in the vascular tree.
• The primary consequence of systemic embolization is ischemic necrosis (infarction) of
downstream tissues, Embolization in the pulmonary circulation leads to hypoxia, hypotension &
right-sided heart failure.
45.
Pulmonary Thromboembolism
• Pulmonaryemboli originate from DVTs
• Incidence is 2-4 per 1000 hospitalized patients.
• In over 95% of cases it originates from thrombi within the deep leg veins proximal to the popliteal fossa
• Embolization from lower leg thrombi is uncommon.
• Movement is usually through the right side of the heart to the pulmonary vasculature.
• Emboli can lodge anywhere depending on size
• PE can occlude the main pulmonary artery, bifurcation of the right & left pulmonary arteries (Saddle
embolus) or pass into the smaller branching arterioles.
• Multiple emboli can occur
• A patient who has had one pulmonary embolus is at increased risk for having more.
• Rarely an embolus passes through an atrial or ventricular septal defect & enters the systemic circulation
(Paradoxical embolism).
46.
Major clinical pathologicfeatures of PE
• Most pulmonary emboli (60%–80%) are small & clinically silent.
• Large embolus blocking a major pulmonary artery can lead to sudden death.
• Embolic obstruction of medium-sized arteries & subsequent rupture of
downstream capillaries rendered anoxic can cause pulmonary hemorrhage.
• Such emboli do not usually cause pulmonary infarction due to dual blood
supply
• Embolism to small end-arteriolar pulmonary branches usually causes infarction
• Multiple emboli can cause pulmonary hypertension & right ventricular failure
(cor pulmonale
47.
Systemic Thromboembolism
Causes
• Most80% arise from intra-cardiac mural thrombi
• Two-thirds are associated with left ventricular infarcts
• 25% with dilated left atria (Secondary to mitral valve disease).
• Aortic aneurysms
• Thrombi overlying ulcerated atherosclerotic plaques
• Fragmented valvular vegetations
• Venous system (Paradoxical emboli);
• 10-15% are of unknown origin.
48.
Fat Embolism
a. Softtissue crush injury or rupture of marrow vascular sinusoids (Long
bone fracture) release microscopic fat globules into the circulation.
b. Fat & marrow embolism occurs in some 90% of individuals with
severe skeletal injuries with < 10% showing any clinical effects
c. Few case develop symptomatic fat embolism syndrome
characterized by pulmonary insufficiency, neurologic symptoms,
anemia, thrombocytopenia & a diffuse petechial rash that is fatal in
10% of cases
49.
Amniotic Fluid Embolism
•Uncommon, grave complication of labor & immediate postpartum period (1 in 40,000 deliveries).
• Mortality rate approaches 80%
• Onset is characterized by:
Sudden severe dyspnea Cyanosis Hypotensive shock
Seizures coma.
• Others are pulmonary edema DIC secondary to release of thrombogenic substances from amniotic fluid..
• The underlying cause is the entry of amniotic fluid into maternal circulation through tears in the
placental membranes and/or uterine vein rupture.
• Histology: Shows squamous cells shed from fetal skin, lanugo hair, fat from Vernix Caseosa, and mucin
derived from the fetal respiratory or gastrointestinal tracts in the maternal pulmonary microcirculation.
Other findings include marked pulmonary edema, diffuse alveolar damage& systemic fibrin thrombi
generated by DIC
50.
Air Embolism
Gas bubbleswithin the circulation can coalesce, obstruct vascular flow & cause
distal ischemic injury.
Causes
• Obstetric or laparoscopic procedures
• Coronary artery bypass surgery or neurosurgery (Cerebral arterial circulation)
Decompression sickness
• Special form of gas embolism caused by sudden changes in atmospheric pressure.
• Seen in Scuba divers, underwater construction workers & persons in unpressurized
aircraft who undergo rapid ascent.
51.
MECHANISM
• When airis breathed at high pressure (Deep sea dive), increased amounts of gas (Particularly nitrogen) become
dissolved in the blood & tissues.
• If the diver then ascends (Depressurizes) too rapidly, the nitrogen expands in the tissues & bubbles out of solution in
the blood to form gas emboli.
• Rapid formation of gas bubbles within skeletal muscles & supporting tissues around joints is responsible for the painful
condition called the bends.
• Gas bubbles in pulmonary vasculature causes edema, hemorrhages & focal atelectasis or emphysema, leading to
respiratory distress called “chokes”.
• Bubbles in the CNS can cause mental impairment & even sudden onset of coma.
• A more chronic form of decompression sickness is called caisson disease: Recurrent or persistent gas emboli in the
bones lead to multifocal ischemic necrosis (Heads femurs, tibiae, humeri are most commonly affected).
• Placing affected persons in a high-pressure chamber to force the gas back into solution treats acute decompression
sickness.
• Subsequent slow decompression permits gradual gas resorption & exhalation so that obstructive bubbles do not re-
form.
52.
INFARCTION
An infarct isan area of ischemic necrosis caused by occlusion of the vascular supply to the affected
tissue.
• Arterial thrombosis or arterial embolism underlies the vast majority of infarction
• Infarction primarily occurs in the heart & the brain (Myocardial or cerebral infarction)
• Others are Kidneys, spleen, pulmonary & bowel infarction, ischemic necrosis of distal extremities
(Gangrene) in diabetics
Less common causes of arterial obstruction include:
• Vasospasm
• Expansion of an atheroma secondary to intra-plaque hemorrhage
• Extrinsic compression of a vessel (Tumour)
• Dissecting aortic aneurysm
• Edema within a confined space (Anterior tibial compartment syndrome).
53.
Uncommon causes oftissue infarction include:
• Vessel twisting (Testicular torsion or bowel volvulus)
• Traumatic vascular rupture
• Entrapment in a hernia sac.
• Note
• Venous thrombosis can cause infarction but the more common outcome is simply congestion.
• Typically bypass channels rapidly open to provide sufficient outflow to restore the arterial
inflow.
• Infarcts caused by venous thrombosis thus usually occur only in organs with a single efferent
vein (Testis or ovary).
•
54.
MORPHOLOGY
Classification of Infarcts
a.According to their colour (Amount of hemorrhage)
• Red (Hemorrhagic)
• White (Pale or anemic)
b. Presence or absence of microbial infection
• Septic
• Bland.
RED INFARCTS
• Occurs as a result of venous occlusions (Ovarian torsion); (2)
• In loose tissues (Lung) where blood can collect in infarcted zones
• Tissues with dual circulations (Lung & small intestine)
• In previously congested tissues (Consequence of sluggish venous outflow)
• Re-established flow after infarction has occurred (Angioplasty of an arterial obstruction).
55.
WHITE INFARCTS
• Occurwith arterial occlusions
• In solid organs
• Organs with end-arterial circulations (Heart, spleen & kidney)
NOTE
• Infarcts tend to be wedge shaped with the occluded vessel at the apex & the organ
periphery forming the base
• The margins of acute infarcts typically are indistinct & slightly hemorrhagic
• Later the edges become better defined by a narrow rim of hyperemia due to inflammation.
• Infarcts resulting from arterial occlusions in organs without dual circulation typically become
progressively paler & more sharply defined with time
56.
• In comparisonhemorrhagic infarcts are the rule in lung & other spongy organs
• In most tissues the main histologic finding associated with infarcts is ischemic
coagulative necrosis
• Most infarcts are ultimately replaced by scar
• The brain is an exception to these generalizations because ischemic tissue
injury in the CNS results in Liquefactive necrosis.
• Septic infarcts occur when infected cardiac valve vegetations embolize or
when microbes seed necrotic tissue. It is later converted into an abscess,
greater inflammatory response, healing by organization & fibrosis.
57.
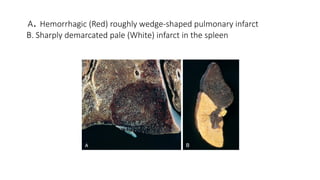
A. Hemorrhagic (Red)roughly wedge-shaped pulmonary infarct
B. Sharply demarcated pale (White) infarct in the spleen
58.
Factors that influenceinfarct development
A. Anatomy of the vascular supply: The presence or absence of an alternative blood supply is the most important
factor in determining whether occlusion of an individual vessel causes damage.
• Lungs: Dual supply by pulmonary & bronchial arteries
• Liver: Hepatic artery & the portal vein
• Hand & forearm: Radial & ulnar arterial supply
• All are resistant to infarction.
• By contrast kidney & spleen both have end-arterial circulations & arterial obstruction generally leads to
infarction.
B. Rate of occlusion: Slowly developing occlusions are less likely to cause infarction because they allow time for
the development of collateral blood supplies. For example, small inter-arteriolar anastomoses interconnect the 3
major coronary arteries.
C. Tissue vulnerability to hypoxia: Neurons undergo irreversible damage when deprived of their blood supply for
only 3 to 4 minutes. Myocardial cells, although hardier than neurons, still die after only 20 to 30 minutes of
ischemia. By contrast, fibroblasts within myocardium remain viable after many hours of ischemia
59.
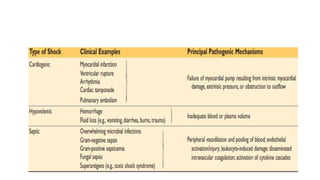
SHOCK
Defined as astate of diminished cardiac output (CO) or reduced effective circulating
blood volume, impairs tissue perfusion leading to cellular hypoxia.
Causes
• Severe hemorrhage Extensive trauma or burns Myocardial infarction
• Pulmonary embolism Microbial sepsis.
CLASSIFICATION
A. Cardiogenic: Results from low cardiac output due to myocardial pump failure.
• Causes
• Myocardial infarction
• Ventricular arrhythmias
60.
• Extrinsic compressioncalled cardiac tamponade
• Outflow obstruction (Pulmonary embolism).
B. Hypovolemic: Results from low cardiac output due to loss of blood or plasma volume (Hemorrhage or
fluid loss from severe burns)
C. Septic shock: From microbial infections, associated with severe systemic inflammatory response
syndrome (SIRS).
Mechanism: Massive outpouring of inflammatory mediators from innate & adaptive immune cells
(Arterial vasodilation, vascular leakage & venous blood pooling) resulting in tissue hypoperfusion, cellular
hypoxia & metabolic derangements (Organ dysfunction, organ failure & death.
D. Neurogenic shock: From loss of vascular tone associated with anesthesia or secondary spinal cord
injury
E. Anaphylactic shock: From systemic vasodilation & increased vascular permeability triggered by an
immunoglobulin E-mediated hypersensitivity reaction
62.
PATHOGENESIS OF SEPTICSHOCK
• Mortality rate ranges between 20-30%.
• Most frequently caused by Gram-positive bacterial infections, Gram-negative bacteria & fungi.
• Following activation, the cells & factors initiate a number of inflammatory responses leading to
septic shock & multiorgan dysfunction
PATHOPHYSIOLOGY OF SEPTIC SHOCK
1. Inflammatory & counter-inflammatory responses
2. Endothelial activation & injury
3. Induction of a procoagulant state
4. Metabolic abnormalities or derangements
5. Organ dysfunction
63.
1. Inflammatory &counter-inflammatory responses. In sepsis, various microbial cell wall constituents
engage receptors on cells of the innate immune system, triggering pro-inflammatory responses.
The initiators of inflammation in sepsis involve the following signaling pathways
• Toll-like receptors (TLRs): Recognize microbe-derived substances containing “Pathogen-associated
molecular patterns” (PAMPs)
• G-protein–coupled receptors: Detect bacterial peptides
• C-type lectin receptors: (Dectins)
• On activation, innate immune cells produce numerous cytokines (TNF, IL-1, IFN-gamma, IL-12, & IL-
18 plus other inflammatory mediators (High-mobility group box 1 protein (HMGB1).
• Reactive oxygen species & lipid mediators like prostaglandins & platelet-activating factor (PAF) are
also elaborated.
• These effector molecules induce endothelial cells (And other cell types) to up-regulate adhesion
molecule expression & further stimulate cytokine & chemokine production.
64.
• The complementcascade is also activated by microbial components, both directly &
through the proteolytic activity of plasmin resulting in the production of:
a. Anaphylatoxins (C3a, C5a)
b. Chemotactic fragments (C5a)
• C. Opsonins (C3b).
• All contributing to the pro-inflammatory state.
• NOTE
• Microbial components can activate coagulation directly through factor XII &
indirectly through altered endothelial function.
• The resulting widespread activation of thrombin may further augment inflammation
by triggering protease activated receptors on inflammatory cells.
65.
Counter-regulatory immunosuppressive mechanisms
•The hyper-inflammatory state initiated by sepsis triggers counter-regulatory
immunosuppressive mechanisms involving both innate & adaptive immune cells.
Mechanisms for immune suppression include
• Shift from pro-inflammatory (TH1) to anti-inflammatory (TH2) cytokines
• Production of anti-inflammatory mediators (Soluble TNF receptor, IL-1 receptor antagonist &
IL-10)
• Lymphocyte apoptosis
• Immunosuppressive effects of apoptotic cells
• Induction of cellular anergy.
• The counter-regulatory mechanisms may occasionally override the inflammatory responses &
the resultant immune suppression renders such patients susceptible to superinfections
66.
2. Endothelial activation& injury
• The pro-inflammatory state & endothelial cell activation associated with
sepsis lead to widespread vascular leakage & tissue edema
• Inflammatory cytokines loosen endothelial cell tight junctions, making
vessels leaky & resulting in the accumulation of protein-rich edema fluid
throughout the body.
• This alteration impedes tissue perfusion which may be exacerbated by
attempts to support the patient with intravenous fluids.
• Activated endothelium also up regulates the production of nitric oxide (NO),
other vasoactive inflammatory mediators (C3a, C5a, & PAF), which may
contribute to vascular smooth muscle relaxation & systemic hypotension
67.
3. Induction ofa procoagulant state
• The derangement in coagulation can lead to DIC
• Sepsis favour coagulation.
• Pro-inflammatory cytokines
• Increase tissue factor production by monocytes & endothelial cells
• Decrease production of endothelial anti-coagulant factors (Tissue factor pathway inhibitor, thrombomodulin
& protein C
• Decrease fibrinolysis by increasing plasminogen activator inhibitor-1 expression
• The vascular leak & tissue edema decrease blood flow at the level of small vessels producing stasis &
diminishing the washout of activated coagulation factors
• The combined effects lead to systemic activation of thrombin & deposition of fibrin-rich thrombi in small
vessels throughout the body further compromising tissue perfusion.
• If DIC is severe, consumption of coagulation factors & platelets leads to their deficiency with concomitant
bleeding & hemorrhage
68.
METABOLIC ABNORMALITIES
a. Septicpatients exhibit insulin resistance & hyperglycemia.
b. Cytokines (TNF, IL-1) stress-induced hormones (Glucagon, Growth hormone & glucocorticoids) &
catecholamine’s all drive gluconeogenesis.
c. Pro-inflammatory cytokines suppress insulin release, promoting insulin resistance in the liver & other
tissues by impairing the surface expression of GLUT-4 (Glucose transporter).
d. Hyperglycemia decreases neutrophil function (Suppressing bactericidal activity), causes increased
adhesion molecule expression on endothelial cells.
e. Initially sepsis is associated with increased glucocorticoid production, later followed by adrenal
insufficiency & a functional deficit of glucocorticoids resulting from depression of the synthetic capacity
of intact adrenal glands or frank adrenal necrosis (DIC) called Waterhouse-Friderichsen syndrome
f. Finally cellular hypoxia & diminished oxidative phosphorylation lead to increased lactate production
and lactic acidosis.
69.
ORGAN DYSFUNCTION
a. Systemichypotension, interstitial edema & small vessel thrombosis all decrease the delivery of
oxygen & nutrients to the tissues (Cellular hypoxia).
b. Mitochondrial damage resulting from oxidative stress impairs oxygen use.
c. High levels of cytokines & secondary mediators diminish myocardial contractility & cardiac output
d. Increased vascular permeability & endothelial injury can lead to acute respiratory distress syndrome
e. Multi-organ failure particularly the kidneys, liver, lungs & heart leading to death.
The severity & outcome of septic shock are dependent on:
a. Extent & virulence of the infection
b. Immune status of the host
c. Presence of other comorbid conditions
d. Pattern & level of mediator production.
70.
Management
• Antibiotics totreat the underlying infection
• Intravenous fluids
• Pressors to maintain blood pressure
• Supplemental oxygen to limit tissue hypoxia.
Superantigens
• A group of secreted bacterial proteins (Superantigens) also cause a syndrome similar
to septic shock (Toxic shock syndrome).
• Superantigens are polyclonal T-lymphocyte activators that induce the release of
high levels of cytokines that result in a variety of clinical manifestations, ranging
from a diffuse rash to vasodilation, hypotension, shock, & death.
71.
Shock is aprogressive disorder that leads to death if the underlying problems are not corrected.
1. An initial non-progressive stage: Reflex compensatory mechanisms are activated & vital organ perfusion is maintained
Mechanisms
a. Various neurohumoral mechanisms help maintain CO & BP
• Baroreceptor reflexes
• Release of catecholamines
• Release of anti-diuretic hormone
• Activation of the renin-angiotensin-aldosterone axis
• Generalized sympathetic stimulation.
b. The net effect is tachycardia, peripheral vasoconstriction & renal fluid conservation
c. Cutaneous vasoconstriction causes the characteristic “Shocky” skin coolness & pallor (Septic shock initially cause cutaneous
vasodilation presenting with warm, flushed skin)
• Coronary & cerebral vessels are less sensitive to sympathetic signals & maintain relatively normal caliber, blood flow & oxygen
delivery
• Thus, blood is shunted away from the skin to the vital organs like the heart & the brain.
• If the underlying causes are not corrected shock then passes to the progressive phase
72.
2. Progressive stage
•Characterized by tissue hypoperfusion & onset of worsening circulatory & metabolic
derangement including acidosis (Widespread tissue hypoxia)
• With persistent oxygen deficit, intracellular aerobic respiration is replaced by anaerobic
glycolysis with excessive production of lactic acid.
• The resultant metabolic lactic acidosis lowers the tissue pH, which blunts the vasomotor
response.
• Arterioles dilate & blood begins to pool in the microcirculation.
• Peripheral pooling worsens the CO putting endothelial cells at risk for the development of
anoxic injury & subsequent DIC.
• With widespread tissue hypoxia vital organs begin to fail.
• In the absence of appropriate intervention the process progresses to an irreversible stage.
73.
3. Irreversible stage
•Cellular & tissue injury is so severe that even if the hemodynamic defects
are corrected survival is impossible.
• Widespread cell injury (Lysosomal enzyme leakage) further aggravating the
shock
• Myocardial contractile function worsens (Increased NO synthesis)
• Ischemic bowel may allow intestinal flora to enter the circulation
(Bacteremic shock superimposed)
• Further progression to renal failure from ischemic injury of the kidney
• Downward spiral finally culminates in death.
74.
MORPHOLOGY
• Hypoxic injuryto cells & tissues from hypoperfusion & microvascular thrombosis.
• Brain, heart, kidneys, adrenals & GIT are most commonly involved.
• Fibrin thrombi seen in tissues more in kidney glomeruli.
• Adrenal cortical cell lipid depletion (Stress) reflecting increased use of stored
lipids for steroid synthesis. Lungs are resistant to hypoxic injury in hypovolemic
shock
• Sepsis or trauma can precipitate diffuse alveolar damage (“shock lung.”)
• Except for neuronal & cardiomyocyte loss, affected tissues can recover
completely if the patient survives.
75.
Clinical Features
• Theclinical manifestations of shock depend on the precipitating insult.
In hypovolemic & cardiogenic shock
• Hypotension
• Weak rapid pulse
• Tachypnea
• Cool, clammy, cyanotic skin. (Septic shock: Skin warm & flushed owing to peripheral vasodilation).
• Primary threat to life is the underlying initiating event (Myocardial infarction, severe hemorrhage,
bacterial infection).
• Worsening renal function can provoke a phase dominated by progressive oliguria, acidosis, and
electrolyte imbalances.
Prognosis varies with the origin of shock & its duration. Better (90%) with hypovolemic & worst with
septic or cardiogenic shock.





























































































































![PERI-PROSTHETIC FRACTURE NAIL-PLATE CONSTRUCT [NPC].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/drarunkumardrmohamedashrafperiprostheticfrasturenail-plateconstructnpc-260209164459-7e9d15a1-thumbnail.jpg?width=640&height=640&fit=bounds)

