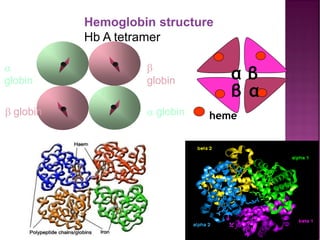
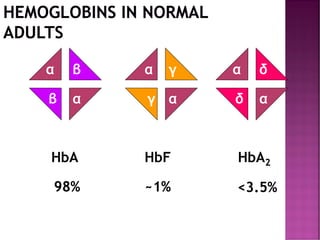
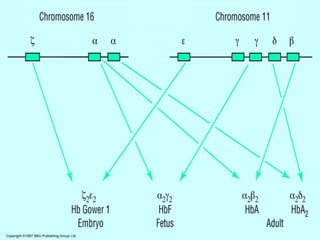
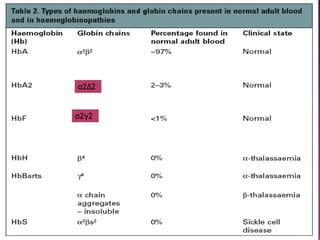
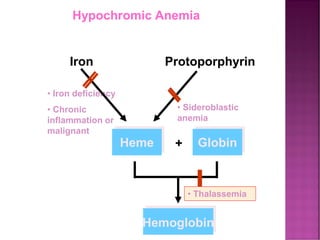
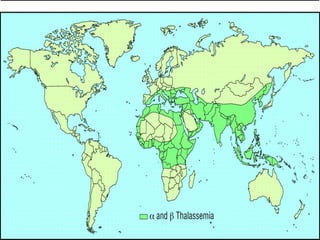
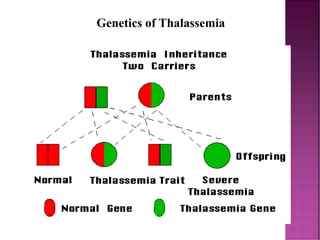
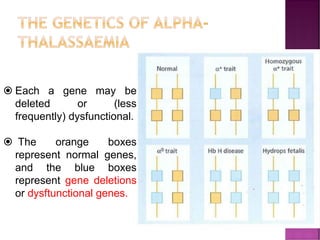
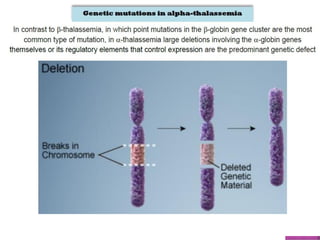
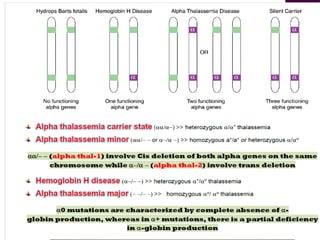
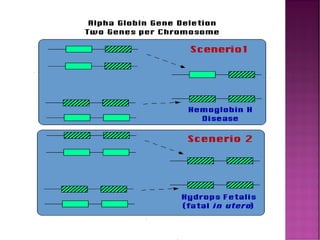
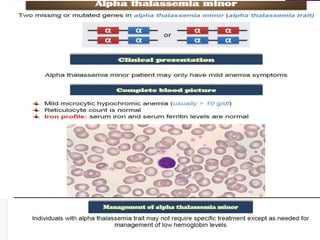
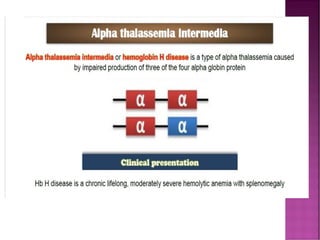
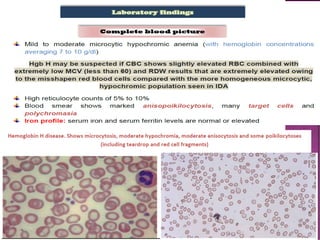
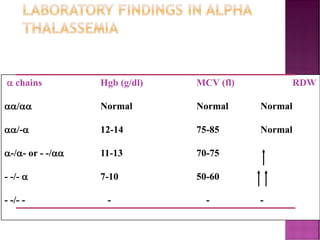
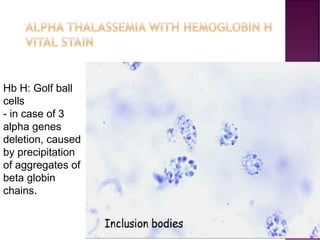
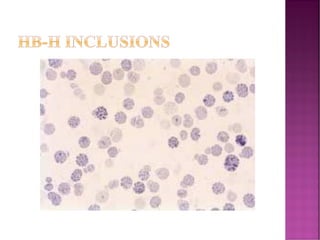
Thalassemias are inherited disorders characterized by deficient synthesis of globin chains, resulting in various forms of microcytic hypochromic anemia. The two main types are alpha and beta thalassemia, differentiated by which globin chain production is reduced. Diagnosis involves clinical features, family history, and hematological tests, with manifestations ranging from asymptomatic carriers to severe transfusion-dependent conditions.