INTRODUCTION TO GLP
•Definition:A quality system concerned with the
organizational process and conditions under which
non-clinical health and environmental safety studies
are planned, performed, monitored, recorded, and
reported.
•Origin: Developed in the 1970s to ensure the
integrity of laboratory studies.
2
3.
IMPORTANCE OF GLP
•Ensures reliability and reproducibility of
data.
• Facilitates regulatory acceptance of study
results.
• Prevents fraudulent practices in research.
4.
GLP VS. GMPVS. GCP
4
Aspect
GLP (Good Lab
Practices)
GMP (Good
Manufacturing
Practices)
GCP (Good
Clinical Practices)
Purpose
Ensures
reliability of non-
clinical studies
Ensures product
quality &
consistency
Ensures ethical &
scientific clinical
trials
Applies to
Preclinical
research
Manufacturing
processes
Human clinical
trials
5.
OECD GLP -OVERVIEW
5
•Organization for Economic Co-operation and
Development (OECD) established international GLP
principles.
•Ensures mutual acceptance of data (MAD) across
member countries.
•Regulates non-clinical safety studies for chemicals,
pharmaceuticals, and pesticides.
6.
6
TYPES OF TESTSCARRIED OUT IN
FACILITIES UNDER GLP
a) The OECD Principles of GLP concern “non-clinical” testing of a chemical or chemical product, examined
under laboratory conditions or in the environment, including work conducted in greenhouses and in the
field. They do not include studies which use human subjects.
b) Examples of studies carried out under GLP include, inter alia:
i) physical-chemical testing;
ii) toxicity studies;
iii) mutagenicity studies;
iv) the environmental toxicity studies on aquatic and terrestrial organisms;
v) studies on behaviour in water, soil and air; bioaccumulation;
vi) studies to determine pesticide residues in food or animal feedstuffs;
vii) studies on effects on mesocosms and natural ecosystems; and
viii) analytical and clinical chemistry testing.
7.
7
TYPES OF CHEMICALSCOVERED BY
THE OECD PRINCIPLES OF GLP TEST
The OECD Principles of GLP apply to the non-clinical safety testing of test items contained in:
i) pharmaceutical products;
ii) pesticide products;
iii) cosmetic product;
iv) veterinary drugs;
v) food additives;
vi) feed additives; and
vii) industrial chemicals.
Depending on the jurisdiction, the Principles of GLP may also be applied to non-clinical safety testing
of other regulated products, such as medical devices.
8.
OECD GLP -COMPLIANCE
MONITORING
• National authorities monitor compliance through
inspections and audits.
• A test facility which has been subject to periodic
inspections by a compliance monitoring
programme (CMP,) and found to be operating in
compliance with the Principles of GLP, is recognised
as a GLP compliant test facility.
• Non-compliance may result in rejection of study
data.
• Ensures reliability for international regulatory
submissions.
8
9.
OECD GLP -MUTUAL ACCEPTANCE OF
DATA (MAD)
• A study conducted under OECD GLP in one member country is
accepted in others.
• Reduces redundant testing and promotes global harmonization.
9
TEST FACILITY ORGANIZATIONAND
PERSONNEL
11
1. Test Facility Organisation and Personnel
•Test Facility Management’s Responsibilities
•Ensure compliance with GLP Principles.
•Identify management roles.
•Provide sufficient resources and qualified personnel.
•Establish and approve Standard Operating Procedures (SOPs).
•Implement a Quality Assurance Program.
•Appoint a Study Director.
•Designate a Principal Investigator if needed for multi-site studies.
•Ensure study plan approval and availability.
•Maintain an SOP historical file and a master schedule.
•Ensure proper characterization of test and reference items.
•Establish procedures for computerized systems.
12.
12
•Study Director’s Responsibilities
•Overallresponsibility for the study and its final report.
•Approve the study plan and amendments.
•Ensure adherence to the study plan and assess deviations.
•Ensure proper documentation and validation of computerized systems.
•Principal Investigator’s Responsibilities
•Conduct delegated phases of the study according to GLP Principles.
•Study Personnel’s Responsibilities
•Be knowledgeable in relevant GLP Principles.
•Comply with study plans and SOPs.
•Record raw data accurately.
•Take health precautions.
13.
13
QUALITY ASSURANCE
PROGRAM
•General
•Establish adocumented Quality Assurance Programme.
•Ensure QA personnel are independent of study conduct.
•Responsibilities of the Quality Assurance Personnel
•Maintain copies of study plans and SOPs.
•Verify study plan compliance.
•Conduct inspections.
•Inspect final reports.
•Report inspection results.
•Prepare a statement for the final report.
14.
14
FACILITIES
•General
•Ensure suitable size,construction, and location.
•Provide adequate separation of activities.
•Test System Facilities
•Assure isolation of test systems and projects.
•Provide rooms for diagnosis, treatment, and control of diseases.
•Include storage rooms for supplies and equipment.
•Facilities for Handling Test and Reference Items
•Provide separate rooms for receipt, storage, and mixing.
•Ensure adequate storage to preserve item integrity and safety.
•Archive Facilities
•Secure storage for study documents and materials.
•Protect contents from deterioration.
•Waste Disposal
•Handle and dispose of waste to maintain study integrity.
15.
15
•Properly locate anddesign apparatus.
•Ensure regular inspection, cleaning, maintenance, and calibration.
•Prevent interference with test systems.
•Label chemicals, reagents, and solutions appropriately.
APPARATUS, MATERIALS AND
REAGENTS
•Physical/Chemical
•Properly locate and design apparatus.
•Ensure integrity.
•Biological
•Maintain proper conditions for biological test systems.
•Isolate new test systems and monitor health.
•Document source, arrival date, and condition.
•Acclimatize test systems.
•Identify test systems properly.
•Clean and sanitize housing.
•Locate field studies to avoid interference.
TEST SYSTEMS
16.
TEST AND REFERENCEITEMS
•Receipt, Handling, Sampling, and Storage
•Maintain records.
•Identify handling, sampling, and storage procedures.
•Label storage containers.
•Characterization
•Identify test and reference items.
•Document identity, purity, and composition.
•Verify test item identity.
•Determine stability.
•Analyze vehicle homogeneity, concentration, and stability.
•Retain a sample for analytical purposes.
17.
STANDARD OPERATING PROCEDURES
(SOPS)
17
•Establishand approve written SOPs.
•Make SOPs readily available.
•Document and acknowledge deviations.
•Cover various test facility activities.
•Test and Reference Items.
•Apparatus, Materials, and Reagents.
•Record Keeping, Reporting, Storage, and Retrieval.
•Test System.
•Quality Assurance Procedures.
18.
18
PERFORMANCE OF THE
STUDY
•StudyPlan:
•A written, dated, and QA-verified plan is required.
•Amendments must be justified and approved.
•Deviations must be documented.
•Short studies may use a general plan with supplements.
•Study Plan Content:
•Include study, item, and sponsor/facility identification.
•Specify dates and test methods.
•Detail test system, administration, design, and records.
•Conduct of the Study:
•Each study must have unique identification for traceability.
•Follow the study plan.
•Record all data directly, promptly, accurately, and legibly.
•Correct data entries clearly, with reasons, dates, and signatures.
•Computerized systems must have full audit trails.
19.
19
REPORTING OF STUDYRESULTS
•General:
•Prepare a final report for each study.
•Signed and dated reports are required from contributing parties.
•The Study Director validates the report.
•Amendments are used for corrections.
•Final Report Content:
•Include study, item, and sponsor/facility identification.
•Specify dates and QA statements.
•Describe materials and methods.
•Present results with analysis and conclusions.
•Indicate where materials are stored.
20.
20
STORAGE AND RETENTIONOF
RECORDS AND MATERIALS
•Retain study plans, data, reports, samples, and other records per regulations.
•Document the final disposition of materials.
•Justify and document early disposal.
•Retain samples/specimens as long as quality permits evaluation.
•Archive materials for organized storage and retrieval.
•Limit archive access to authorized personnel; record all material movement.
•Transfer archives to sponsors if the facility closes.
21.
21
GLP GUIDELINES ACROSS
COUNTRIES
•Different regulatory agencies enforce GLP
standards globally.
• Countries follow OECD GLP or have their national
guidelines.
22.
REGULATIONS SPECIFIC TOUS
• Food and drug administration- 21 CFR part 58: This part prescribes good
laboratory practices for conducting nonclinical laboratory studies that
support or are intended to support applications for research or
marketing permits for products regulated by the Food and Drug
Administration, including food and color additives, animal food
additives, human and animal drugs, medical devices for human use,
biological products, and electronic products
• Environmental protection agency- 40 CFR part 160: This part prescribes
good laboratory practices for conducting studies that support or are
intended to support applications for research or marketing permits for
pesticide products regulated by the EPA
23.
23
GLP REGULATIONS BYCOUNTRY
Country Regulatory Authority Unique GLP Requirements
India
National GLP Compliance
Monitoring Authority (NGCMA)
Requires periodic inspections,
certification by NGCMA, and
mandatory adherence for
studies supporting regulatory
submissions.
USA
FDA (21 CFR Part 58) & EPA (40
CFR Part 160)
GLP regulations cover
pharmaceuticals, pesticides,
and environmental testing
with strict data integrity and
archiving requirements.
UK
Medicines and Healthcare
products Regulatory Agency
(MHRA)
Requires pre-study inspections
and adherence to UK-specific
guidelines alongside OECD
GLP.
Australia
National Association of Testing
Authorities (NATA)
Recognizes OECD GLP but
mandates NATA accreditation
for test facilities.
Japan
Ministry of Health, Labour and
Welfare (MHLW) & PMDA
Enforces additional
documentation and local
language requirements for
GLP submissions.
Canada Health Canada
Aligns with OECD GLP but
includes specific requirements
for environmental and
24.
24
MOST COMMON NONCOMPLIANCE
OBSERVATIONSFROM GLP STUDIES
• 21 CFR 58.33(b): Study director failed to assure that all experimental data were accurately
recorded and verified
• 21 CFR 58.35(b)(5): QAU failed to determine that protocol and SOP deviations were made
without proper authorization and documentation
• 21 CFR 58.35(b)(6): QAU did not review the final report
• 21 CFR 58.51: Archives
• 21 CFR 58.81(b): SOPs have not been established
• 21 CFR 58.113(a)(1): Formulation analysis
25.
25
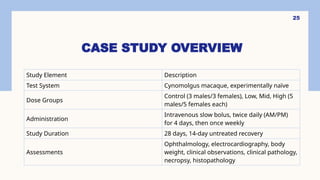
CASE STUDY OVERVIEW
StudyElement Description
Test System Cynomolgus macaque, experimentally naïve
Dose Groups
Control (3 males/3 females), Low, Mid, High (5
males/5 females each)
Administration
Intravenous slow bolus, twice daily (AM/PM)
for 4 days, then once weekly
Study Duration 28 days, 14-day untreated recovery
Assessments
Ophthalmology, electrocardiography, body
weight, clinical observations, clinical pathology,
necropsy, histopathology
26.
26
KEY EVENTS (DAYS2-3)
Study Day Event
Day 2
High dose females showed clinical signs of
toxicity after AM dose, worsening after PM
dose. No signs in males or lower dose
females.
Day 3 (AM)
High dose females showed more severe
toxicity. High dose males showed minimal
signs. Lower dose groups and controls
remained normal. Affected animals showed
signs of dehydration and renal impairment.
Day 3 (Noon)
Study Director stopped dosing high dose
females, created a Protocol Amendment, and
notified staff.
All animals except high dose females were
27.
27
PROTOCOL AMENDMENT ANDSTUDY
CONTINUATION
Study Day Event
Day 4
All animals except high dose females dosed
per protocol. Sponsor and Study Director
signed the Protocol Amendment, and it was
distributed.
Days 5-28 & Recovery
Remaining animals dosed per protocol. High
dose males showed minimal toxicity, but
recovered. Renal impairment observed in mid-
and high-dose animals.
Necropsy & Histopathology
No test article-related findings at necropsy.
Histopathology showed dose-dependent renal
epithelial cell damage.
28.
28
• In thestudy described, did a protocol deviation occur?
A. No, the Protocol Amendment covered the change in dosing
B. No, the Study Director email covered the change in dosing
C. Yes, the change in dosing was not covered by the Protocol
Amendment
D. No, the stopped dosing was due to animal welfare
29.
29
• Correct Answer:C. Yes, the change in dosing was not
covered by the Protocol Amendment.
• Explanation:The dosing change occurred before the
Protocol Amendment was finalized and distributed. This
constitutes a deviation from the approved protocol.
30.
30
REGULATORY OBSERVATION
• Observation:The quality assurance unit failed to determine
whether any deviations from approved protocols or
standard operating procedures had been made without
proper authorization and documentation.
• Regulation Cited: 21 CFR 58.35(b)(5)
• Implication: This highlights the importance of proper
documentation and QAU oversight in GLP studies to ensure
protocol adherence.
31.
31
CONCLUSION
• GLP ensuresthe integrity of non-clinical safety data.
• OECD GLP principles promote global harmonization.
• Compliance is essential for regulatory approvals and scientific credibility.
32.
32
GLP CHALLENGES ANDFUTURE
TRENDS
• Increasing complexity in testing methodologies.
• Integration of AI and automation in GLP studies.
• Stricter compliance monitoring and enforcement.




























































![Hypothalamus short notes on location, function and disorders by Dr. Neha [PT]...](https://cdn.slidesharecdn.com/ss_thumbnails/hypothalamusbydr-260124142231-2b48143d-thumbnail.jpg?width=640&height=640&fit=bounds)


















