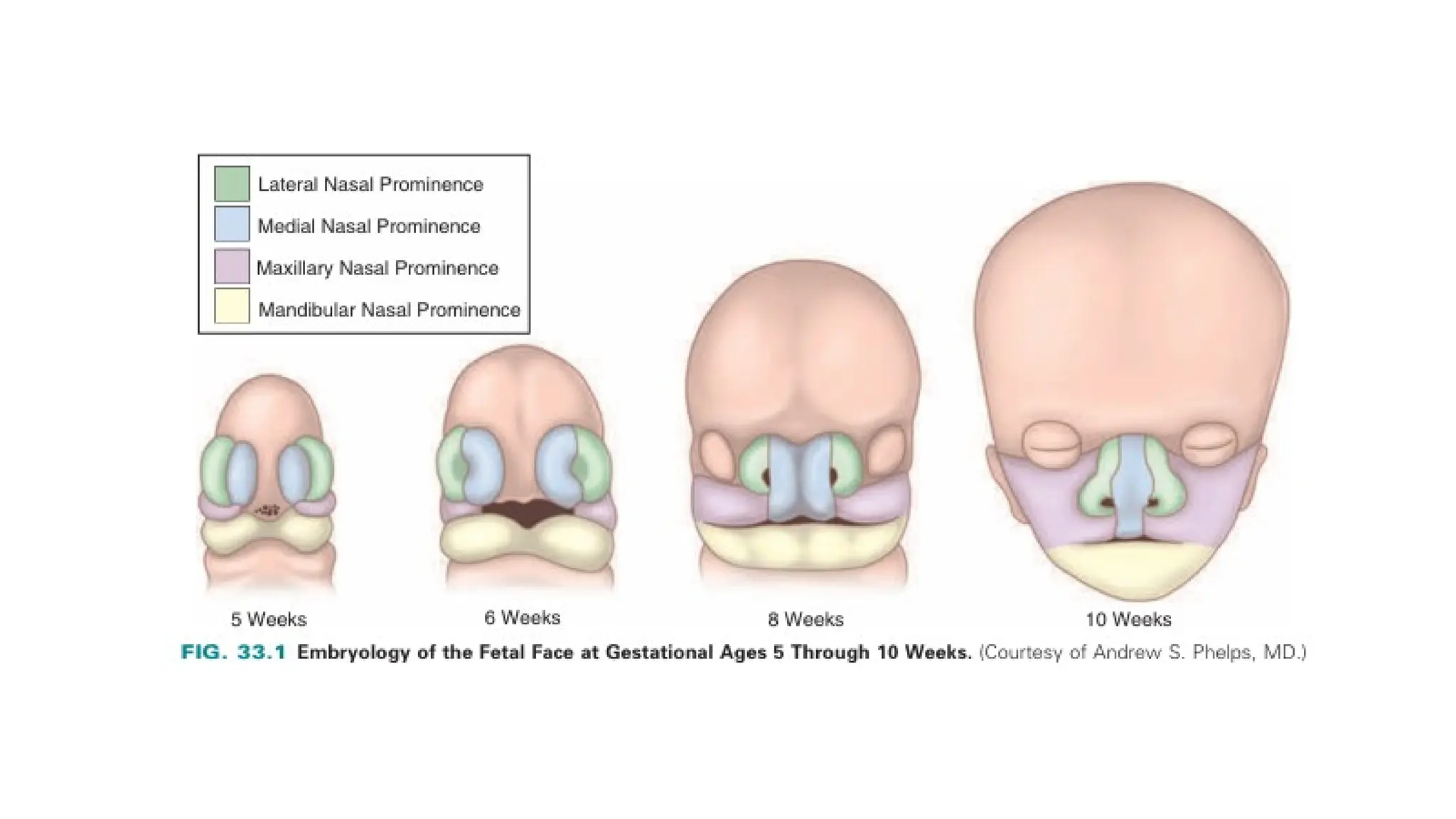
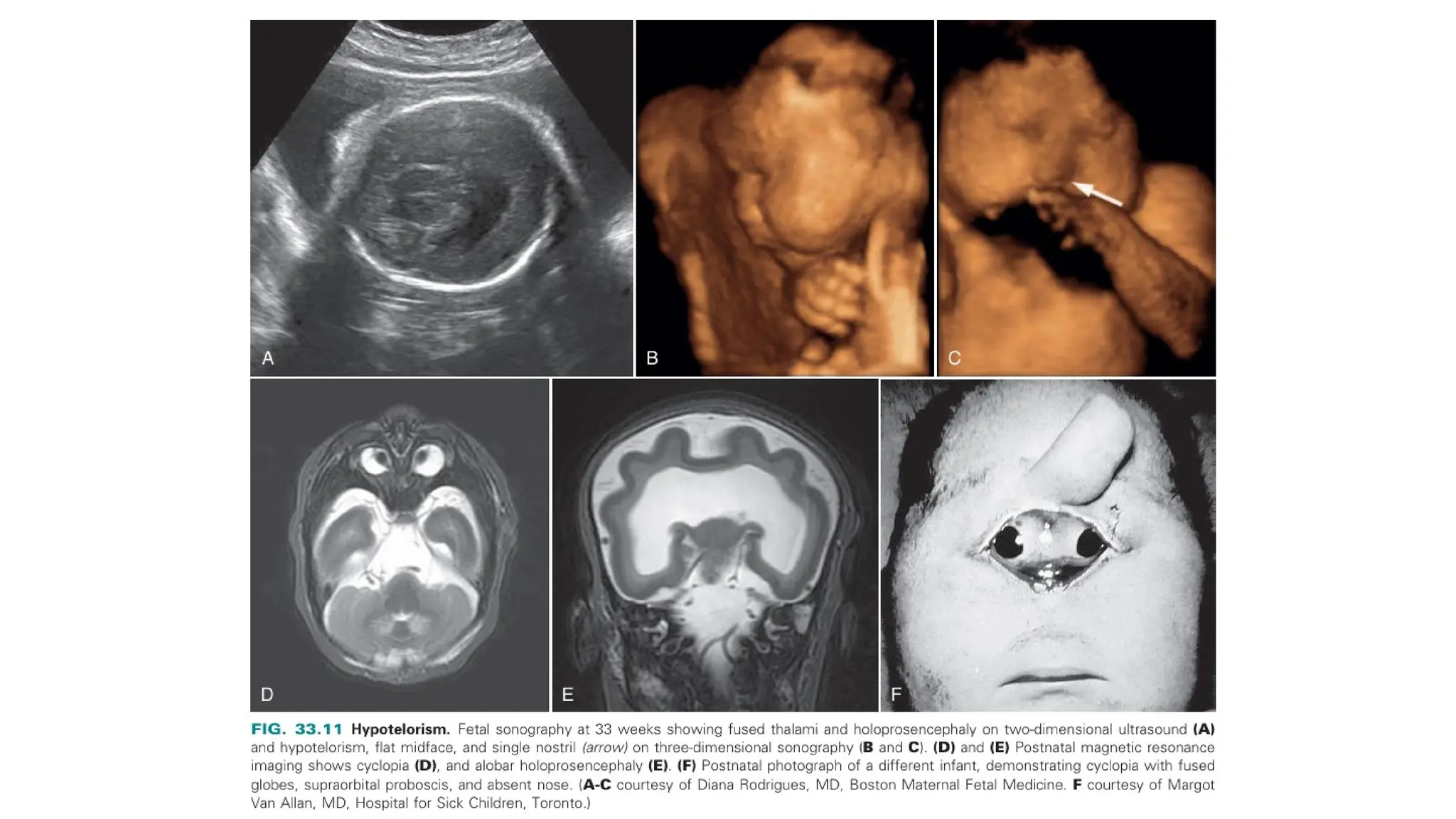
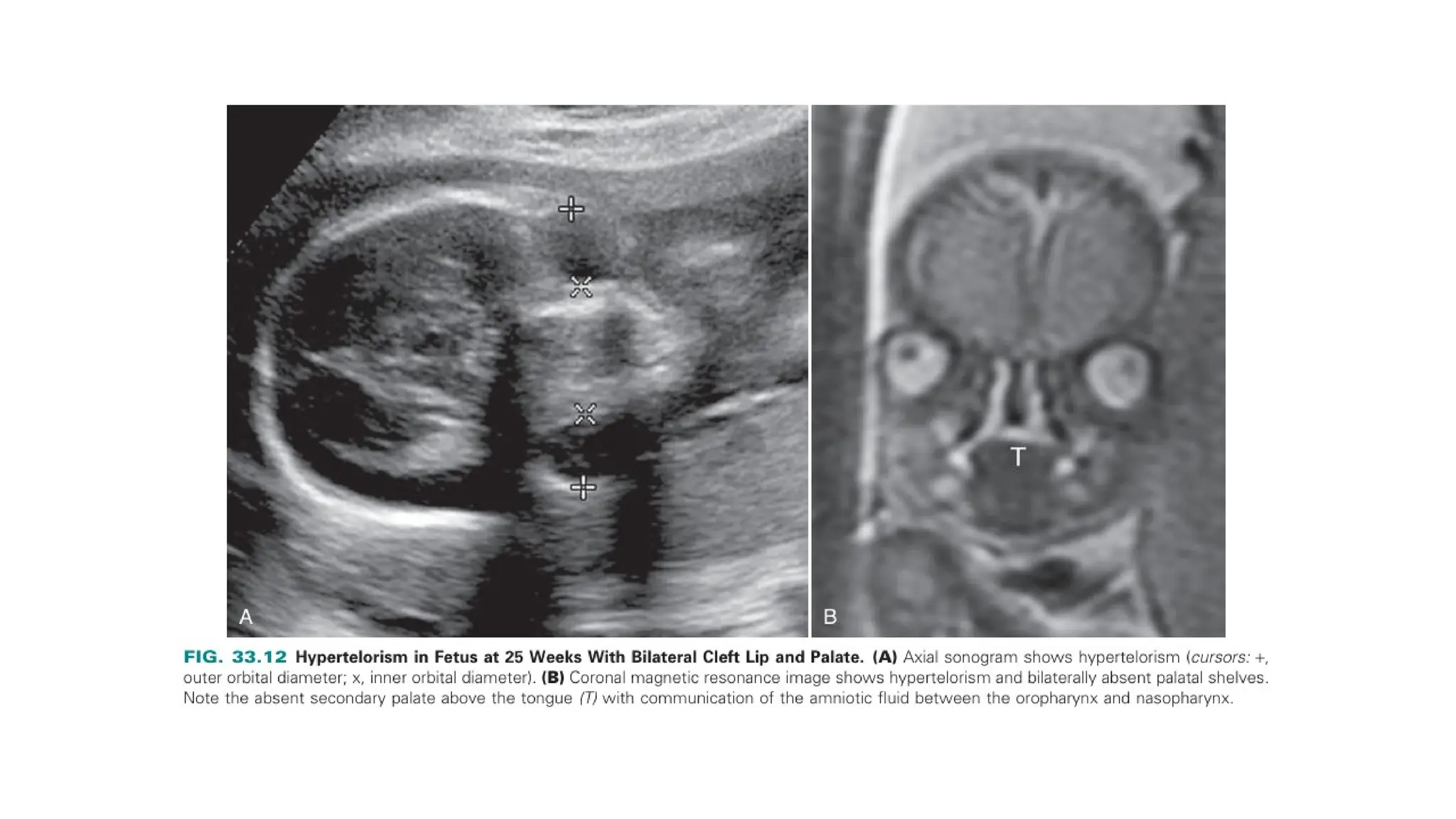
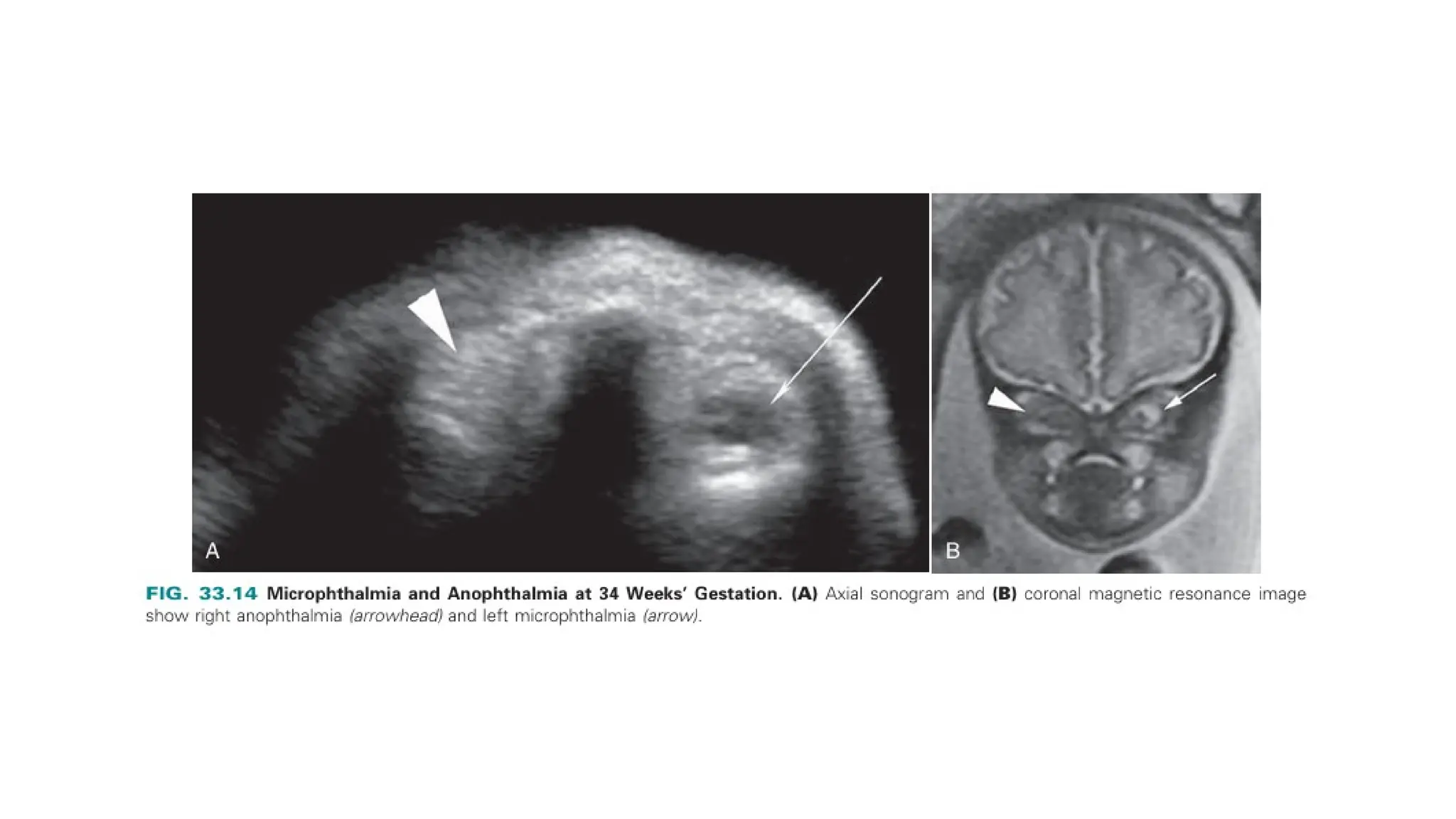
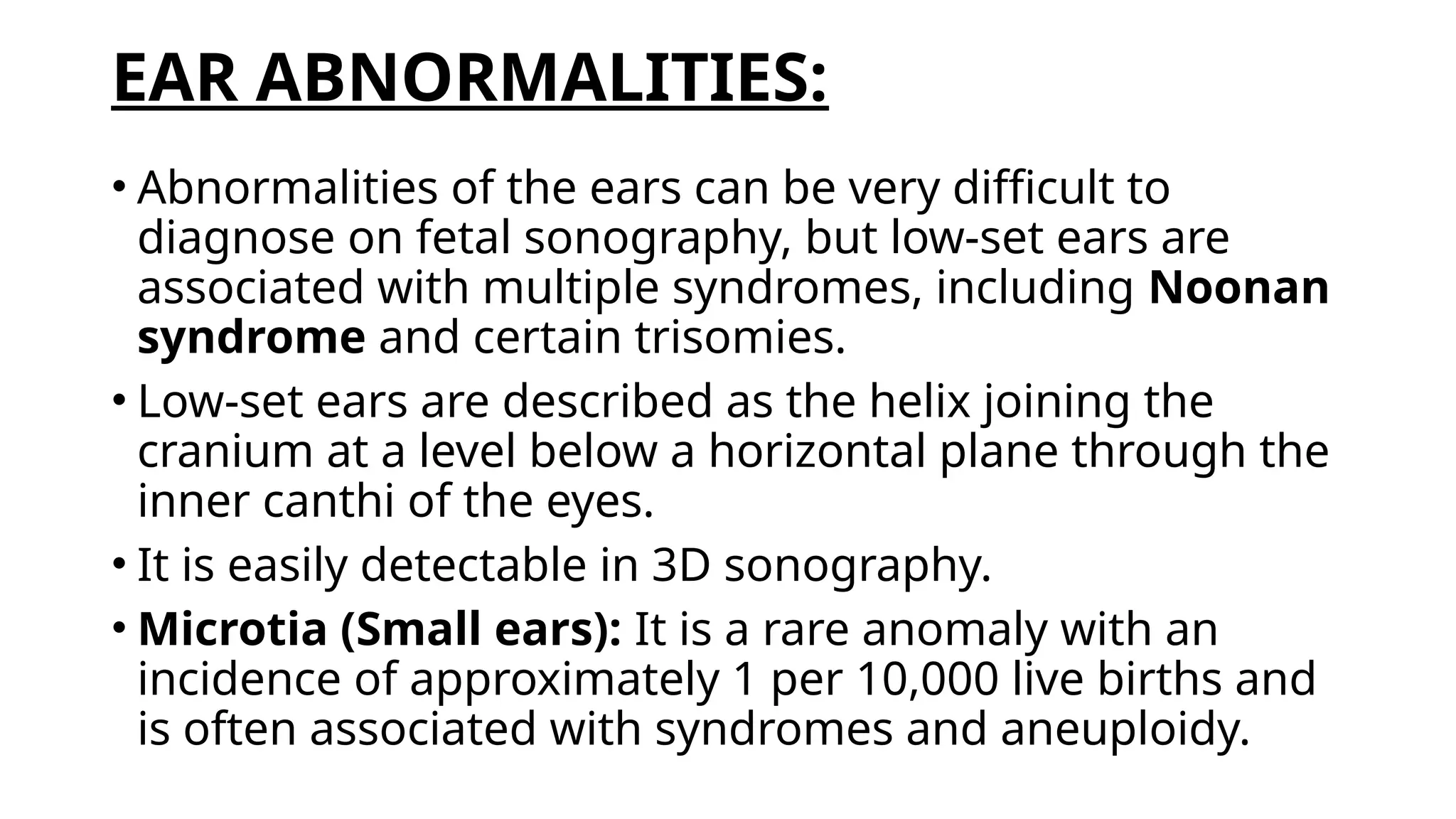
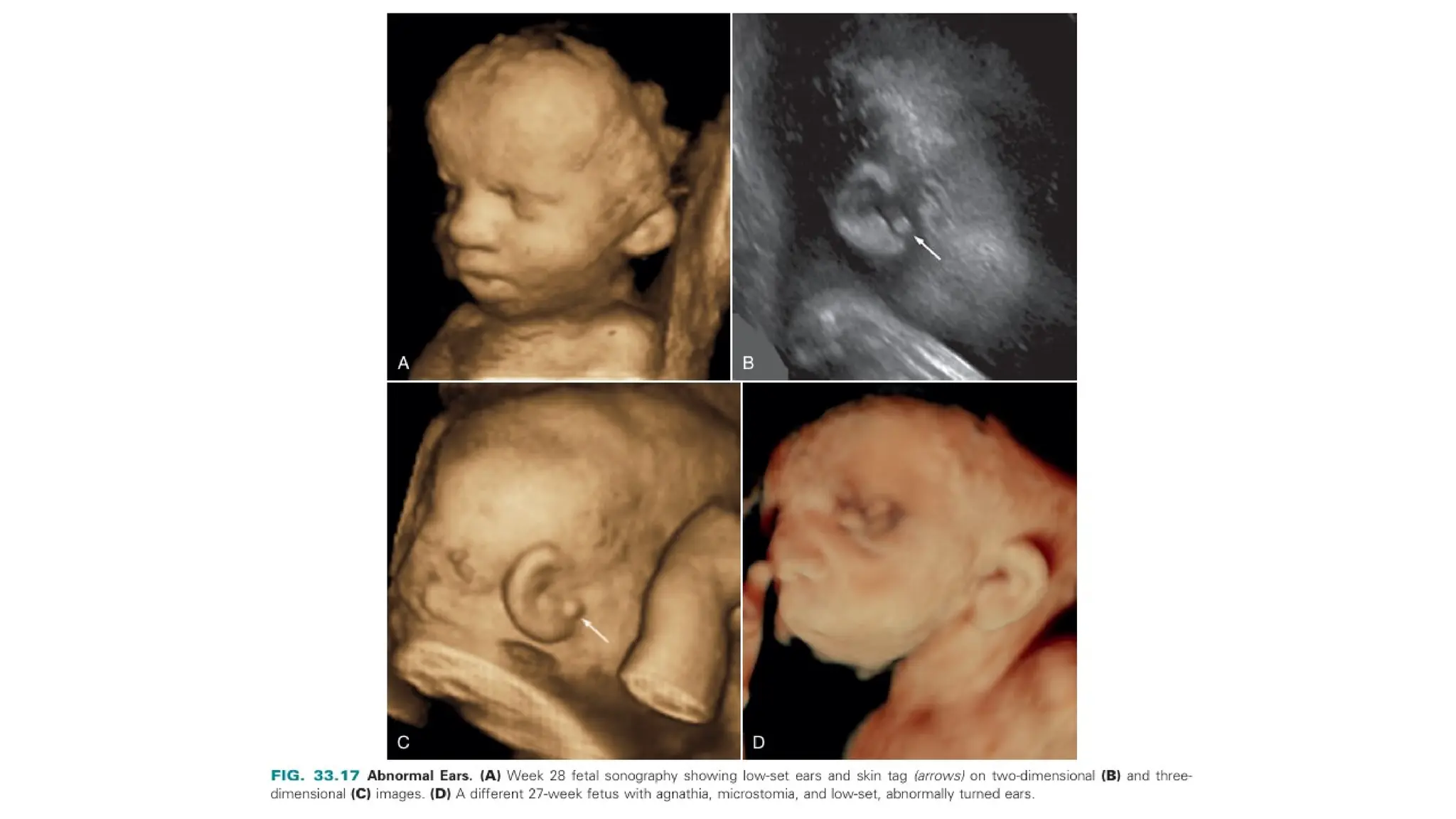
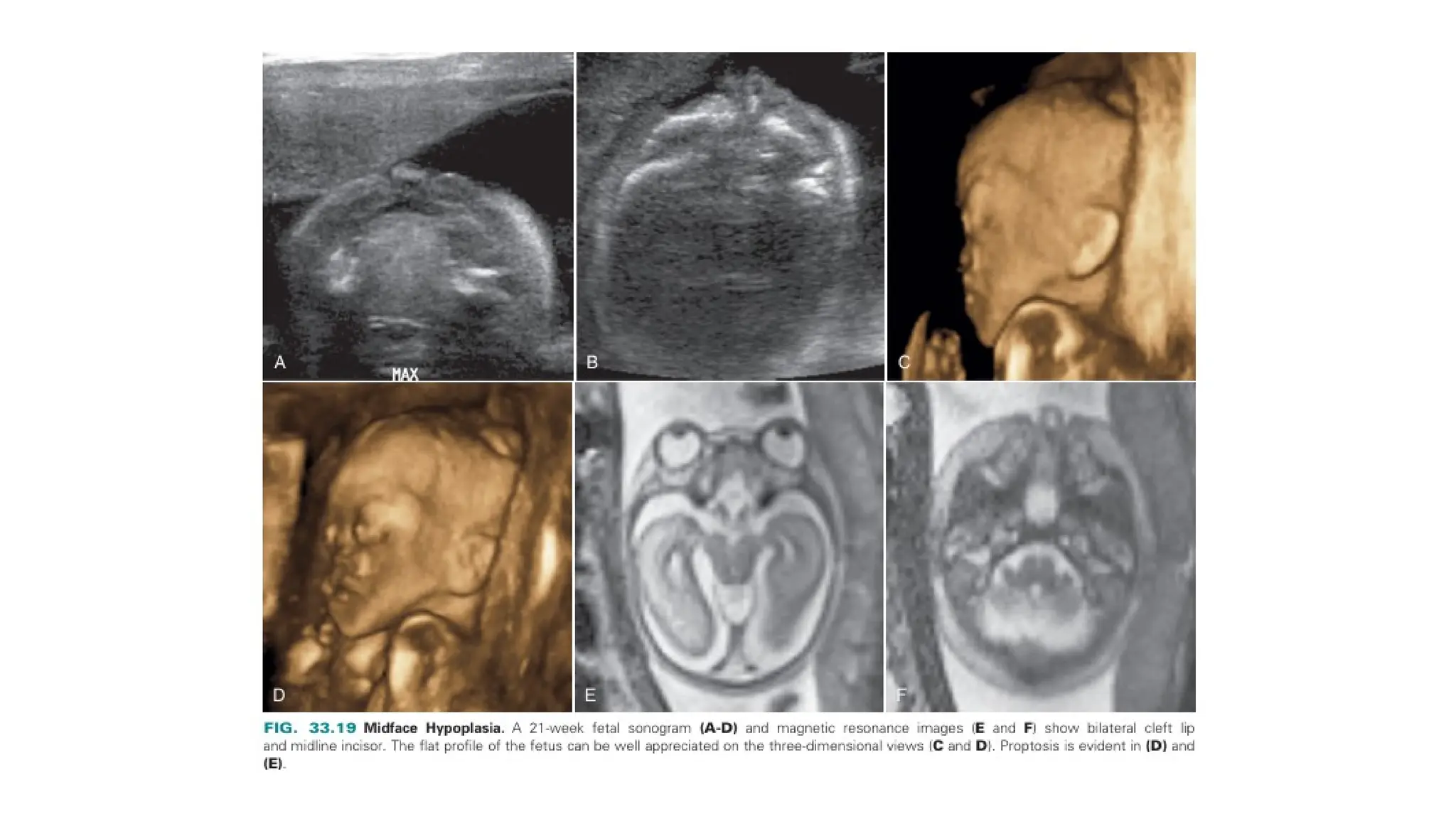
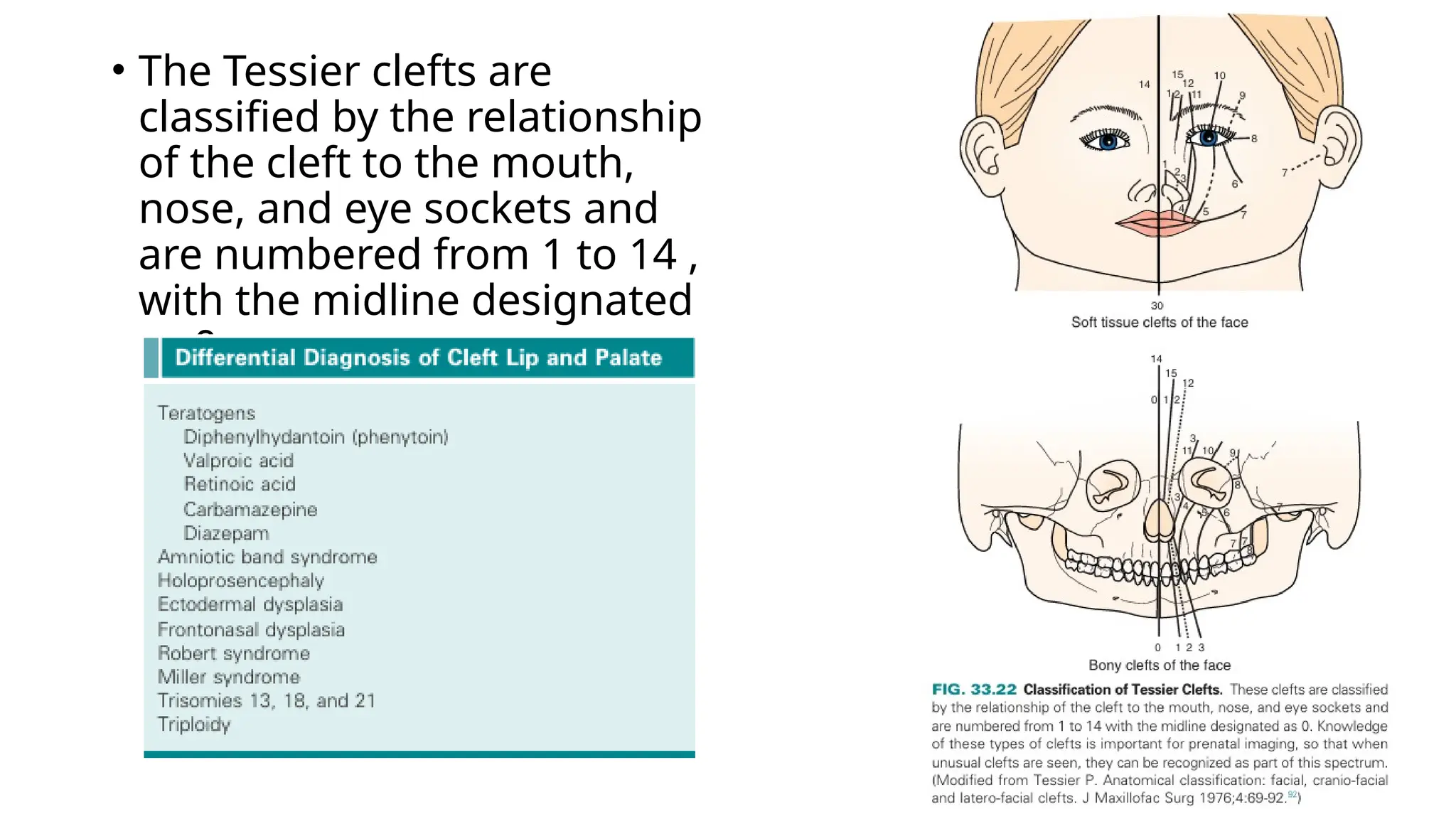
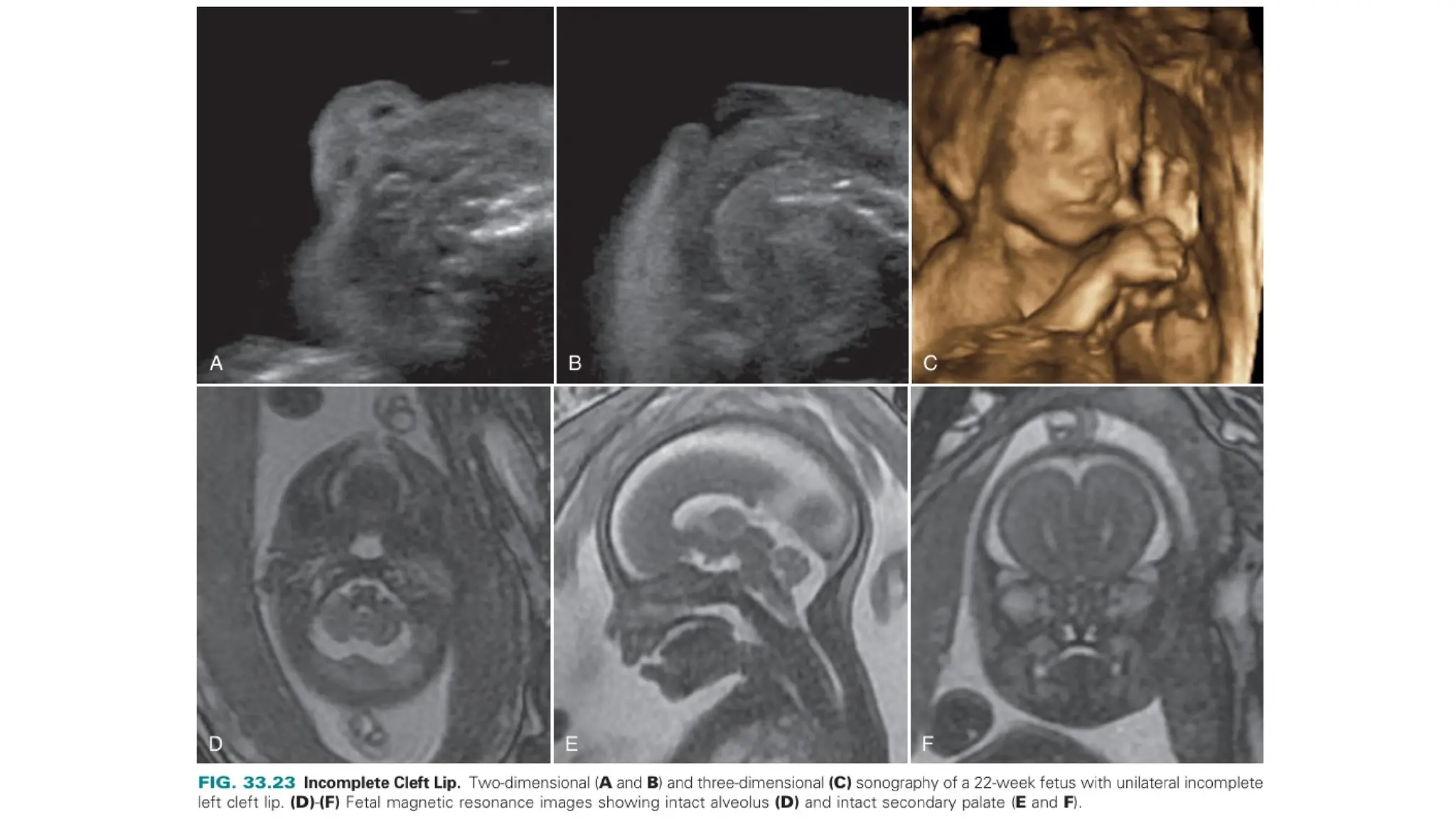
The document discusses the embryonic development and abnormalities of the fetal head and neck, detailing the formation of facial structures and potential conditions like clefts, malformations, and tumors. Key features including ultrasound diagnostics for various abnormalities, such as hypotelorism, coloboma, and congenital cataracts, are highlighted. It also covers the development of the neck, including lymphatic malformations, teratomas, and goiter, with implications for prenatal diagnoses and associated syndromes.