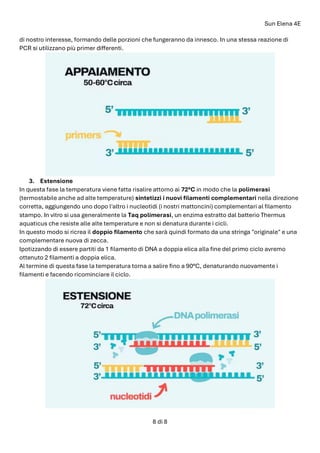
Il documento descrive il processo di DNA fingerprinting, utilizzando marcatori STR per analizzare e confrontare profili genetici al fine di determinare relazioni di parentela. Vengono spiegate tecniche come l'elettroforesi e la PCR, evidenziando l'importanza degli enzimi di restrizione e dei primers nella generazione di copie di DNA. La procedura include la preparazione del gel e l'analisi dei risultati per confermare la compatibilità genetica tra il figlio e i potenziali padri.