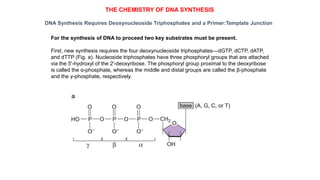
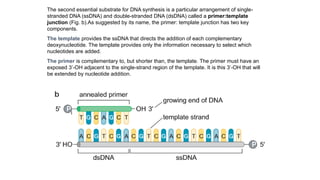
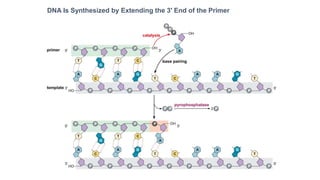
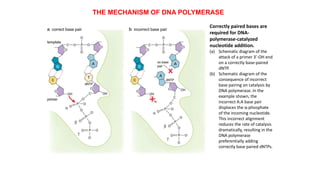
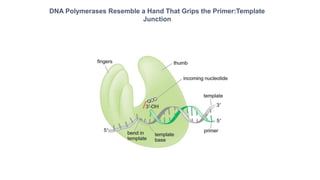
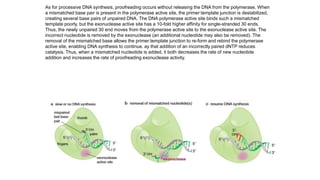
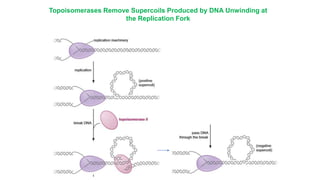
The document summarizes an experiment conducted by Meselson and Stahl to determine the mechanism of DNA replication. They grew E. coli in media containing either normal nitrogen-14 or heavy nitrogen-15. After shifting the cells between the two media for multiple generations, they analyzed the DNA densities.
After one generation, a single band appeared midway between labeled and unlabeled DNA, ruling out conservative replication but not distinguishing between semiconservative and dispersive replication. After two generations, two bands appeared in a 1:1 ratio as predicted by semiconservative replication, but not by dispersive replication. The experiment provided strong evidence that DNA replication occurs through a semiconservative mechanism.




![The results of one more round of DNA replication
ruled out the dispersive hypothesis.
Dispersive replication would give a product with one-
fourth 15N and three-fourths 14N after two rounds of
replication in a 14N medium.
Semiconservative replication would yield half of the
products as H/L and half as L/L. In other words, the
hybrid H/L products of the first round of replication would
each split and be supplied with new, light partners,
giving the 1:1 ratio of H/L to L/L DNAs.
Three replication hypotheses. The conservative
model (a) predicts that after one generation equal
amounts of two different DNAs (heavy/heavy [H/H] and
light/light [L/L]) will occur. Both the semiconservative (b)
and dispersive (c) models predict a single band of DNA
with a density halfway between the H/H and L/L
densities. Meselson and Stahl’s results confi rmed the
latter prediction, so the conservative mechanism was
ruled out. The dispersive model predicts that the DNA
after the second generation will have a single density,
corresponding to molecules that are 25% H and 75% L.
This should give one band of DNA halfway between the
L/L and the H/L band. The semiconservative model
predicts that equal amounts of two different DNAs (L/L
and H/L) will be present after the second generation.
Again, the latter prediction matched the experimental
results, supporting the semiconservative model.](https://image.slidesharecdn.com/dnareplication1para-240412042912-7e3f5426/85/DNA-replication-process-fundamental-pptx-5-320.jpg)