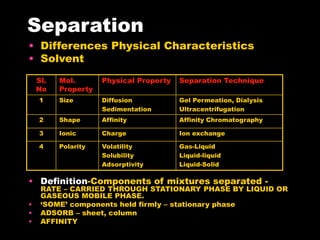
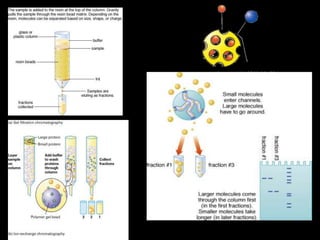
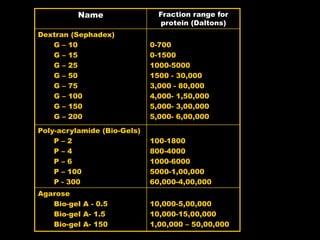
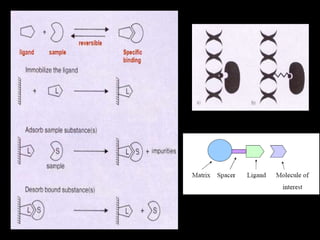
Chromatography is a separation technique developed in the early 20th century for isolating mixtures based on their physical characteristics, using methods such as paper, thin layer, column, and affinity chromatography. Each method utilizes different stationary phases and mobile phases to separate components based on properties like size, shape, ionic charge, and polarity. Applications range from chemical analysis to protein purification, and the technique has evolved significantly since its inception, enabling various industrial and research uses.