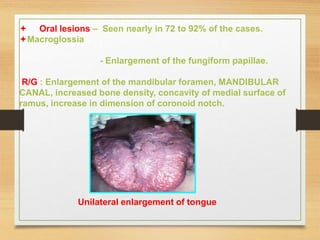
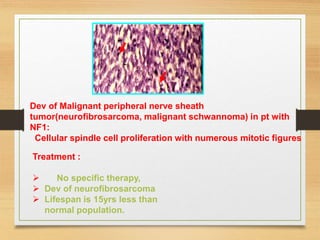
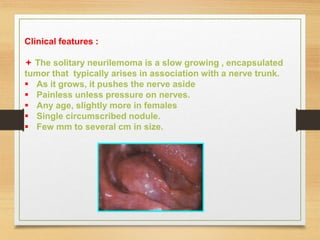
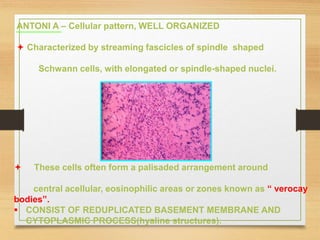
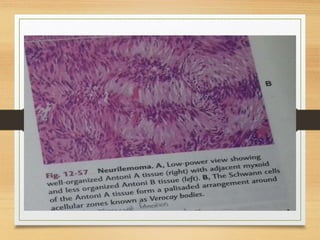
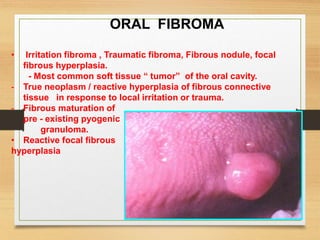
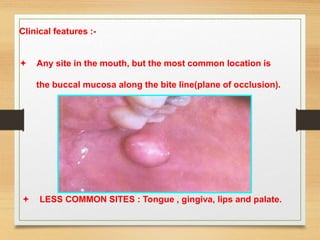
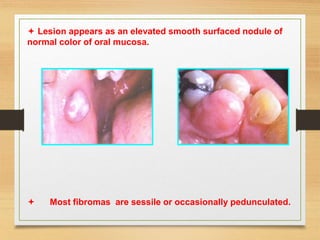
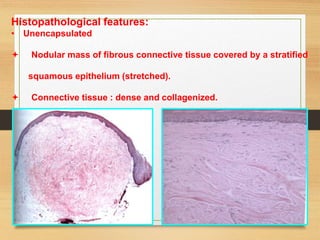
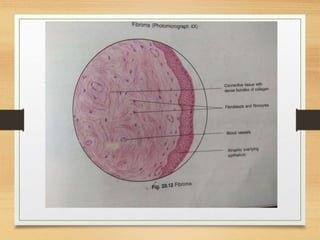
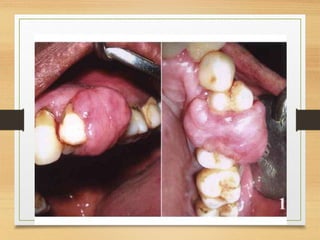
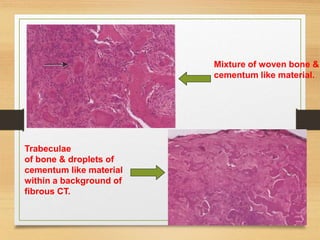
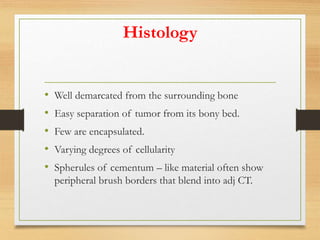
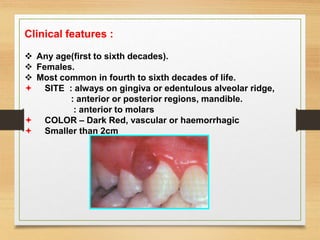
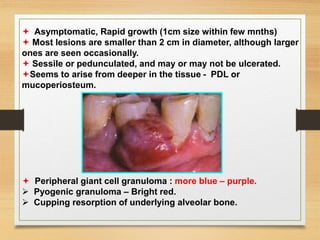
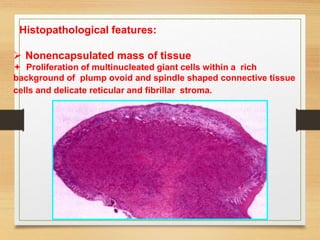
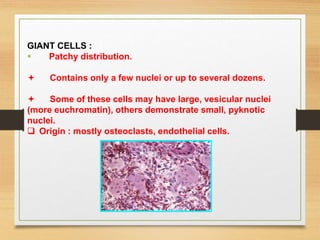
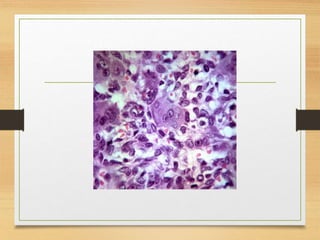
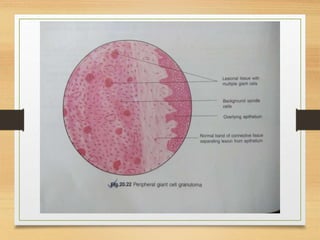
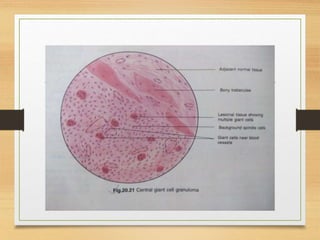
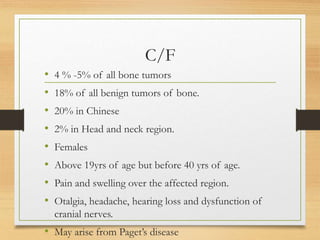
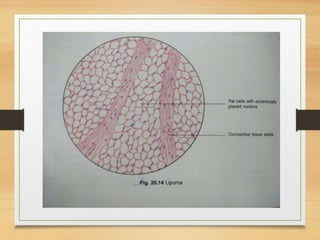
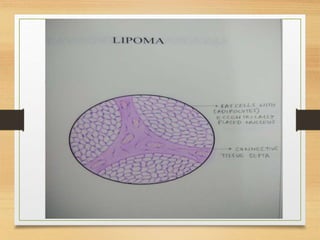
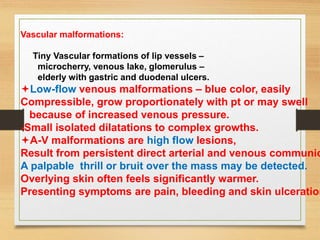
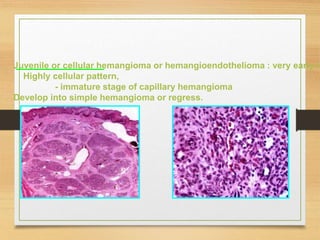
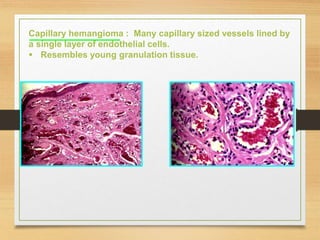
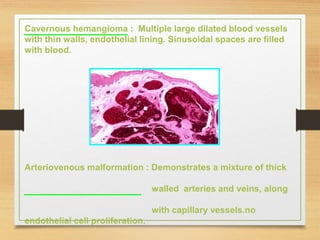
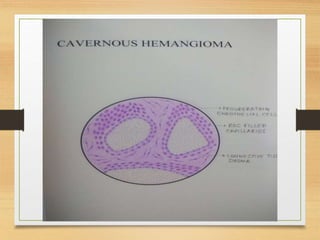
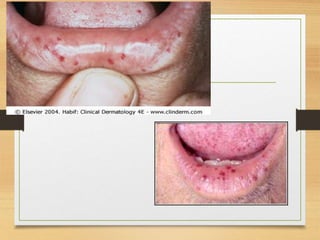
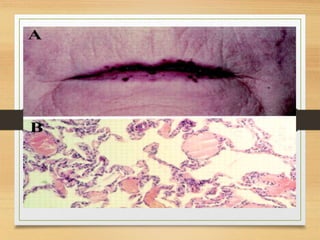
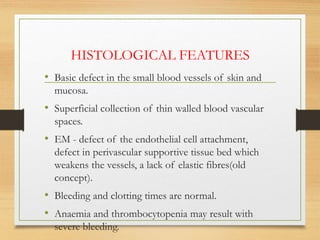
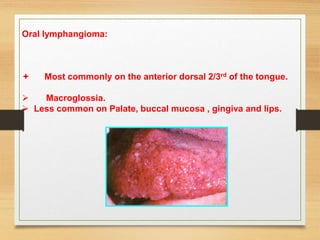
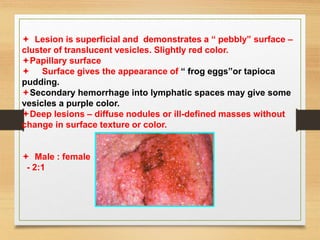
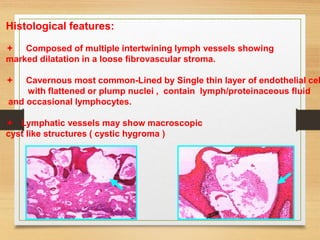
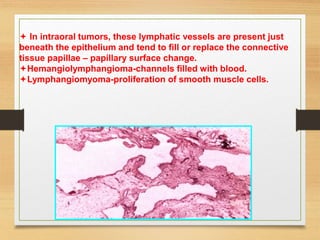
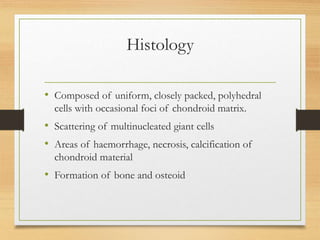
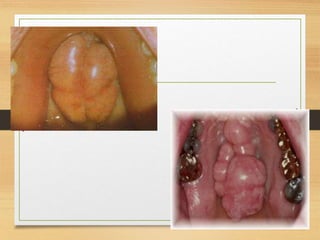
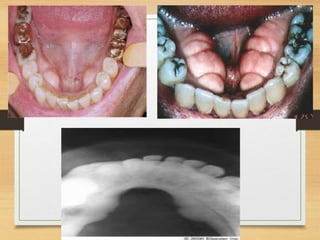
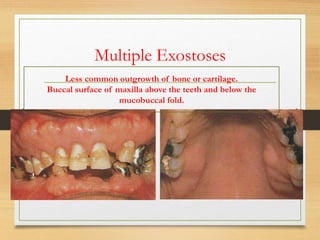
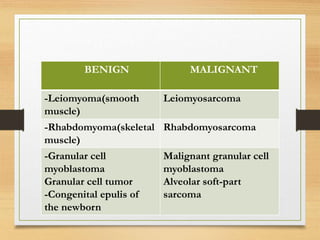
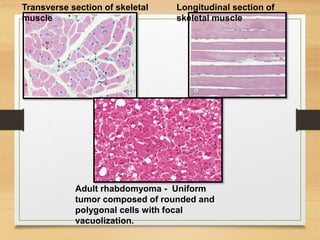
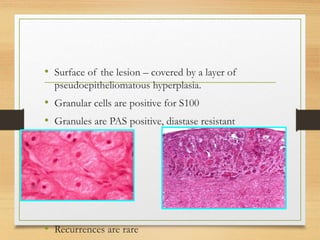
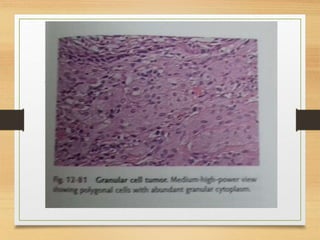
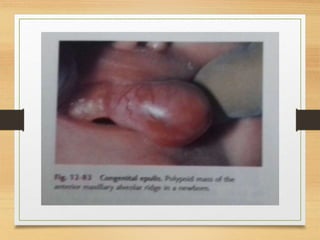
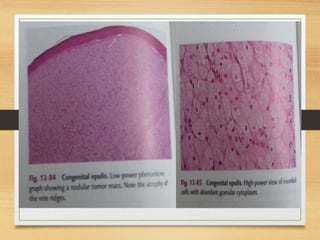
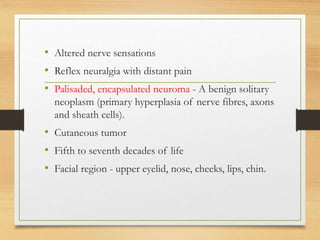
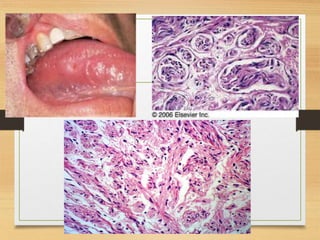
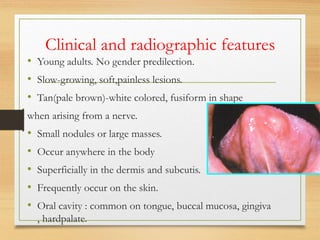
The document discusses non-epithelial tumors of the oral cavity, focusing on connective and soft tissue tumors such as oral fibromas, peripheral ossifying fibromas, and various types of giant cell granulomas. It highlights the differences between benign and malignant tumors, various clinical features, histopathological characteristics, and treatment options for these tumors. The text offers detailed information on common reactive lesions, their clinical presentations, treatment methods, and recurrence rates.






















































































































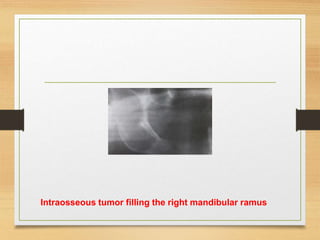

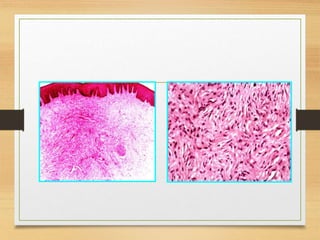









![ The tumors may be present at birth, but they often begin to appear
during puberty and may continue to develop slowly throughout
adulthood. Accelerated growth in pregnancy.
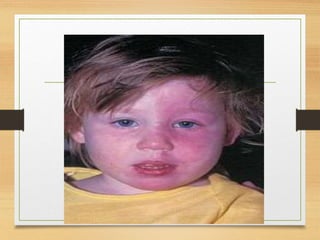
“ Café au lait” [ coffee with milk ] pigmentation on the skin.
SPOTS ARE Smooth-edged, yellow-tan to dark-brown macules
(1to2mm or several cm).
Mostly present at birth or dev in first year of life.
Café au lait pigmentation on the arm](https://image.slidesharecdn.com/connectivetissuetumors-benign-240617171427-2b29497c/85/Connective-tissue-tumors-Benign-for-3rd-bds-ppt-184-320.jpg)
![ “ Axillary freckling” [ Crowe’s Sign ] is also seen.
Freckle: flat , small , light brown spots.
“ Lisch nodules” translucent, brown pigmented spots on the
iris are found in nearly all affected individuals.
- HYPERTENSION IS A COMMON PROBLEM.
- Pheochromocytomas(tumor of adrenal glands)
Other possible abnormalities are C. N. S. tumors, macrocephaly
mental deficiency, seizures, short stature and scoliosis.](https://image.slidesharecdn.com/connectivetissuetumors-benign-240617171427-2b29497c/85/Connective-tissue-tumors-Benign-for-3rd-bds-ppt-185-320.jpg)