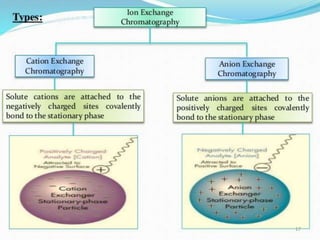
The document provides a comprehensive overview of chromatography and related separation techniques, detailing methods such as extraction, crystallization, distillation, and filtration. It explores the principles and history of chromatography, emphasizing its effectiveness in separating complex mixtures and applications across various scientific fields. Additionally, it elaborates on affinity chromatography, including ligands, immobilization techniques, and the importance of matrix properties for efficient separation.