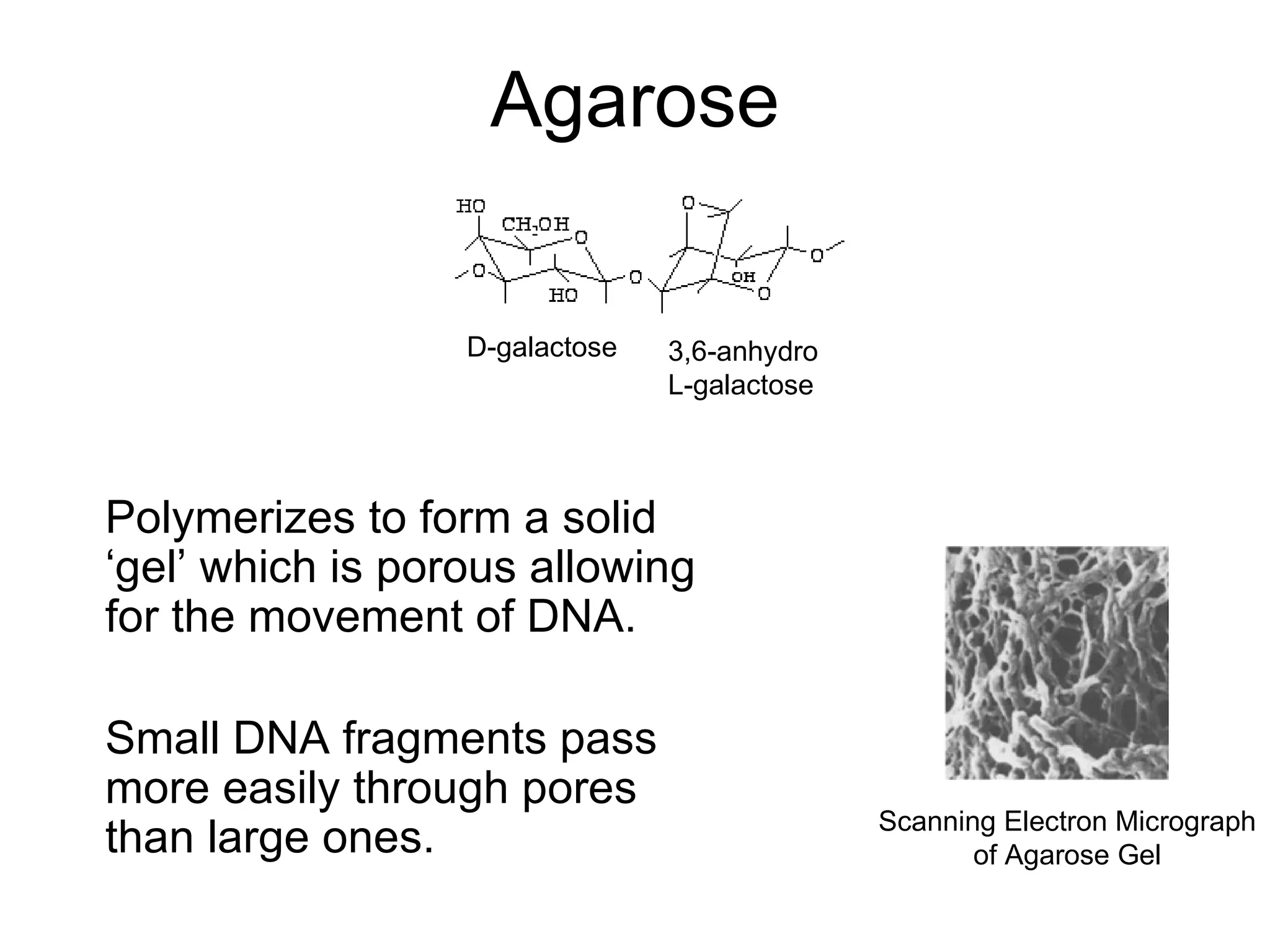
The document discusses DNA isolation and agarose gel electrophoresis. It describes how DNA can be extracted from various biological sources and purified. The process involves lysing cells, removing proteins, and separating DNA from other molecules. Agarose gel electrophoresis is then used to separate DNA fragments by size, as smaller fragments move more quickly through the gel under an electric field. The document outlines the basic steps for preparing an agarose gel, loading samples mixed with dye, running the gel, and visualizing DNA bands under UV light.