Prostaglandins:
PGs are biologicallyactive derivatives of 20 carbon atoms polyunsaturated essential
fatty acids that are released from cell membrane phospholipids . They are the major
lipid derived autocoids ( eicosanoids)
biosynthesis of prostaglandins
3.
Prostaglandin receptors
• Prostaglandinsbring about their effects by acting on prostanoid receptors , which
are all G –protein coupled receptors . Some of them act through cAMP and others
through a inositol triphosphate pathway.
• There are five classes of prostanoid receptors . They are
• DP (for PGD2) – subtypes DP1 and DP2
• EP ( for PGE2) –subtypes EP1 to EP4
• FP ( for PGF2α )
• IP ( for PGI2)
• TP ( for TXA2)
4.
Physiological and pathologicalrole
1. CVS
PGs do not circulate in blood and have no role in regulating systemic vascular
resistance. However, PGI2 generated in the vascular endothelium, mainly by
COX-2, appears to be involved in the regulation of local vascular tone as a dilator.
PGE2 is continuously produced locally in the ductus arteriosus by COX-2 during
foetal life—keeps it patent; at birth its synthesis stops and closure occurs . These
PGs may also be important in maintaining placental blood flow.
PGs, generated mainly by COX-2, along with LTs and other autacoids may
mediate vasodilatation and exudation at the site of inflammation
5.
2. Platelets
TXA2 producedby platelets and PGI2 produced by vascular endothelium probably
constitute a mutually antagonistic system: preventing aggregation of platelets while
in circulation and inducing aggregation on injury, when plugging and thrombosis are
needed.
TXA2 produced by platelet COX-1 plays an important role in amplifying
aggregation
3. uterus
Foetal tissues produce PGs. At term PGF2α has been detected in maternal blood. It is
postulated that PGs mediate initiation and progression of labour.
Because PGs are present in high concentration in semen, it is believed that they so
coordinate movements of the female genital tract that transport of sperms and
fertilization is facilitated.
Dysmenorrhoea in many women is associated with increased PG synthesis by the
endometrium. This apparently induces uncoordinated uterine contractions which
compress blood vessels → uterine ischaemia → pain.
6.
4. Bronchial muscles
PGF2α,PGD2 and TXA2 are potent bronchoconstrictors (more potent than histamine) while
PGE2 is a powerful bronchodilator. PGI2 produces mild dilatation.
Asthma may be due to an imbalance between constrictor PGs (F2α , PGD2 , TXA2 ) on one
hand and dilator ones (PGE2 , PGI2 ) on the other.
5. GIT
PGs may be involved in mediating toxin induced increased fluid movement in secretory
diarrhoeas.
PGs appear to play a role in the growth of colonic polyps and cancer.
PGE2 markedly reduces acid secretion in the stomach. Volume of juice and pepsin content
are also decreased. Release of gastrin is suppressed. The gastric pH may rise upto 7.0.
PGI2 also inhibits gastric secretion, but is less potent. Secretion of mucus and HCO3 ¯ by
gastric mucosal epithelial cells as well as mucosal blood flow are increased. Thus, PGs are
antiulcerogenic
7.
6. kidney
PGE2and PGI2 produced mainly by COX-2 in the kidney appear to function as
intrarenal regulators of blood flow as well as tubular reabsorption in kidney.
Renin release in response to sympathetic stimulation and other influences may be
facilitated by PGs.
Bartter’s syndrome, characterized by decreased sensitivity to angiotensin II is
associated with increased PG production
7. CNS
PGE2 may mediate pyrogen induced fever and malaise. Pyrogens, including
cytokines released during bacterial infection, trigger synthesis of PGE2 in the
hypothalamus, which resets the thermostat to cause fever.
PGs may be functioning as neuromodulators in the brain by regulating neuronal
excitability. A role in pain perception, sleep and some other functions have been
suggested
8.
8. ANS
PGs maymodulate sympathetic neuro transmission in the periphery
9. Peripheral nerves
PGs appear to serve as algesic agents during inflammation. They cause tenderness and
amplify the action of other algesics.
10. Eye
Locally produced PGs appear to facilitate aqueous humor drainage
11.Endocrine system
PGE2 facilitates the release of anterior pituitary hormones—growth hormone, prolactin,
ACTH, FSH and LH as well as that of insulin and adrenal steroids.
9.
12. Metabolism
PGEs areantilipolytic, exert an insulin like effect on carbohydrate metabolism and
mobilize Ca2+ from bone. They may mediate hypercalcaemia due to bony metastasis.
Opioid autocoids
Opioid autocoids refer to naturally occurring molecules in the body that mimic the effects of
opioids by binding to opioid receptors
➤ Opioid is a psychoactive chemical that works by binding to opioid receptors which are
found principally in the CNS, PNS and GIT.
➤ The receptors in these organ system mediate both the benificial and side effects of opioids.
➤ Opioids divide into two types- endogenous and exogenous. Some endogenous opioids that
bind to the receptors are enkephalins, endorphins, endomorphins, dynorphins, and
nociception/orphanin
➤ Exogenous opioids like morphine, heroin, and fentanyl are substances that are introduced
into the body and bind to the same receptors as the endogenous opioids.
10.
Five types ofopioid receptors have been discussed- mu(MOR),Kappa(KOR),delta( DOR) ,
nociception (NOR) , zeta (ZOR)
➤ Within these different types are a subset of subtypes, mu1, mu2, mu3, kappa1, kappa2,
kappa3, delta1, and delta2.
➤ Mu1,2,3 receptors (MOR) bind to endogenous ligands - beta-endorphin, Endomorphin 1
and 2.
➤ The mu-1 receptor is responsible for analgesia and dependence.
The mu-2 receptor is vital for euphoria, dependence, respiratory depression, miosis,
decreased digestive tract motility/constipation
➤ Mu-3 receptor causes vasodilation
➤ Kappa receptors (KOR) bind to dynorphin A and B. They provide analgesia, diuresis
➤ Delta receptors (DOR) bind to enkephalins . They play a role in analgesia and reduction in
gastric motility.
➤ Nociceptin receptors (NOR) bind to nociceptin/orphanin causing analgesia
➤ Zeta receptors (ZOR) regulate developmental events in a variety of normal and
tumorigenic tissues and cells.
11.
Physiological role ofopioid autocoids-
a.Analgesia- Endogenous and exogenous opioid autacoids cause analgesia.
b. MOR play a central role in reducing the stress response through inhibition of secretion of
norepinephrine.
c. Mood and reward- Presence of high density of MOR in the limbic system(emotional center)
regulates mood and rendering these receptors potential targets to treat mood disorders like
anxiety and depression. MOR stimulation, results in the release of dopamine. Dopamine is
responsible for the rewarding effects produced by opioid administration.
Pathological actions of opioid autocoids:
a. Respiratory Depression- Opioid receptors are abundant in the respiratory center.
Stimulation of these receptors leads to irregular and slow breathing.
b. Constipation- Opioid receptor activation by agonists causes slowing of propulsive motility of
the gut mediated through inhibition of acetylcholine.
c. Orthostatic Hypotension and Syncope- Opioid receptors are present in cardiac tissue; their
activation leads to hyperpolarization of membranes and activation of the vagus nerve.These
changes result in peripheral vasodilation and bradycardia, which ultimately causes low BP
12.
d.Endocrine abnormalities-
Stimulation ofopioid receptors located in the hypothalamus inhibits GnRH
release, which results in reduced estrogen and testosterone secretion . Hence,
chronic activation of these receptors leads to osteoporosis and sexual dysfunction,
presenting as infertility and increased bone fragility
e. Immune Dysfunction- Opioid receptors are present on immune cells, namely
natural killer (NK) cells, and phagocytes, and their stimulation leads to repression
of their activity resulting in blunting of the immune response and delayed wound
healing
f. Sleep Changes- Activation of opioid receptors in the reticular formation alters
normal sleep pattern. Opioid agonists through stimulation of these receptors
increase the duration of light sleep, consequently decreasing deep and REM sleep
duration.
g. Mood Changes- Chronic stimulation of MOR in the neurons of hippocampal
region leading to mood dysregulation.
13.
Anti-histamines
Anti histamines arethe drugs that block the action of histamine on histamine receptors.
Conventional anti-histamines are the ones which competitively block H1 receptor.
Classification
I. H1 receptor antagonists
a. First generation antihistamines(sedatives)
Eg. Diphenhydramine, dimenhydrinate, promethazine, pheniramine, chlorpheniramine,
cyclizine , meclizine, buclizine, mepyramine, tripelennamine
b. Second generation (non sedative) anti- histamines
Eg. Loratadine, desloratadine, cetirizine, levocetirizine , fexofenadine, acrivastine ,
azelastine, mizolastine .
II. H2 receptor antagonists
Eg. Cimetidine , ranitidine, famotidine , roxatidine, nizatidine
14.
III. H3 receptorantagonists
Eg . Thioperamide , clobenpropit
IV. H4 receptor antagonists
Eg . Toreforant
H1 receptor antagonists
Actions
Qualitatively all H1 anti-histaminics have similar actions, but there are quantitative
differences, especially in the sedative property.
1. Antagonism of histamine-
• They effectively block the histamine induced bronchoconstriction , contraction of intestinal
and other smooth muscles and triple response –especially wheal , flare , itch
15.
• Fall inBP produced by histamine is blocked
• Release of Adrenaline from adrenal medulla in response to histamine is abolished.
• Constriction of larger blood vessel by histamine is also antagonized.
• Action of histamine on gastric secretion is singularly not affected by these drugs.
2. Anti-allergic action
• Many manifestations of immediate hypersensitivity (type I reactions) are suppressed.
• Urticaria, itching and angioedema are well controlled.
• Anaphylactic fall in BP is only partially prevented.
3.CNS
• Antihistamines produce variable degree of CNS depression. This appears to depend on the
compound’s ability to penetrate the blood-brain barrier and its affinity for the central H1
receptors.
16.
• Individual susceptibilityto different agents varies considerably. The same drug and dose
may incapacitate some subjects, while others may remain alert.
• Some individuals also experience stimulant effects like restlessness and insomnia. Excitement
and convulsions are frequently seen at toxic doses.
• The second generation anti-histaminics are practically non-sedating
• first generation anti-histamines like promethazine , diphenhydramine and meclizine prevent
motion sickness and vomiting due to labyrinthine disturbances.
• Promethazine also controls vomiting of pregnancy and other causes.
• Promethazine and few other anti-histaminics reduce tremor, rigidity and sialorrhoea of
parkinsonism.
4. Anticholinergic actions
• Many of the first generation H1 blockers have anticholinergic properties. This accounts for both
useful and adverse effects
• Such antihistaminics have antisecretary (used in rhinorrhea) but they also have anticholinergic
side effects like urinary retention and dryness of the mouth.
17.
• Second generationagents do not block the muscarinic receptors
5. Local anaesthetic
• Some drugs like pheniramine, promethazine, diphenhydramine have strong while others
have weak membrane stabilizing property. However, they are not used clinically as local
anaesthetic because they cause irritation when injected s.c.
Mechanism of action
• H1 antihistamines work by acting as inverse agonists at the histamine receptors ,
stabilizing them in their inactive state
• They also work by competitively inhibiting histamine from binding to H1 receptors ,
thereby reducing the effects of histamine on tissues , including reducing allergic
symptoms like itching , runny nose , and sneezing
18.
pharmacokinetics
• The conventionalH1 antihistaminics are well absorbed from oral and parenteral routes,
metabolized in the liver and excreted in urine.
• They are widely distributed in the body and enter brain. The newer compounds penetrate
brain poorly accounting for their low/absent sedating action.
• Duration of action of most agents is 4–6 hours, except meclozine, chlorpheniramine,
mesolastine, loratadine, cetirizine and fexofenadine which act for 12–24 hours or more.
Adverse effects
Side effects of first generation H1 antihistaminics are frequent, but generally mild.
Individuals show marked differences in susceptibility to side effects with different drugs.
Some tolerance to side effects develops on repeated use.
Sedation, diminished alertness and concentration, light headedness, motor incoordination,
fatigue and tendency to fall asleep are the most common.
19.
• Few individualsbecome restless, nervous and are unable to sleep. Second generation
compounds are largely free of CNS effects
• Dryness of mouth, alteration of bowel movement, urinary retention and blurring of vision
can be ascribed to anticholinergic property.
• Epigastric distress and headache may be felt
• Local application can cause contact dermatitis
• Acute overdose produces central excitation, tremors, hallucinations, muscular
incordination, convulsions, flushing, hypotension, fever and some other features of
belladonna poisoning.
Contraindications
1. Drugs that can produce sedation and CNS depression like alcohol , barbiturates ,
clonidine, benzodiazepines should not be combined with sedative antihistaminics because
sedation gets added up
2. They should not be given with antimuscarinics because the effects get added up
20.
Therapeutic uses
1. Allergicreactions
• Useful for the prevention and treatment of symptoms of allergic reactions .
• They are effective in allergic rhinitis , allergic conjunctivitis , hay fever , urticaria , pruritis,
some allergic skin rashes and pollinosis
2. Common cold
• First generation agents reduce rhinorrhea and afford symptomatic relief in common cold
because of their antimuscarinic properties
3. Motion sickness
• Given 30 min before travelling , antihistamines prevent motion sickness –promethazine ,
dimenhydrinate , meclizine, cyclizine are used
21.
4. Vertigo
• Usefulin treating vertigo and other vestibular disturbances –dimenhydrinate , meclizine,
cinnarizine are preferred
• Cinnarizine is the H1 antihistamine having additional anticholinergic, anti-5-HT, sedative
and vasodilator properties which has been widely used in vertigo.
5. Antiemetic
• Promethazine is used to prevent drug induced and post-operative vomiting
• Some of them have also been used in morning sickness – particularly doxylamine
6. Preanesthetic medication
• For sedative , anticholinergic , and antiemetic properties promethazine has been used as
preanesthetic medication
22.
7. As sedative,hypnotic, anxiolytic
• Antihistamines with CNS depressant action have been used as sedative and to induce sleep,
especially in children
• Hydroxyzine has been used as an anxiolytic
8. Parkinsonism
• Diphenhydramine, orphenadrine and promethazine are useful in drug induced
parkinsonism due to their anticholinergic action
9.Cough
• Antihistaminics like chlorpheniramine, diphenhydramine and promethazine are
constituents of many popular cough remedies. They have no selective cough suppressant
action, but may afford symptomatic relief by sedative and anticholinergic property
23.
10. Acute muscledystonia
• Caused by anti dopaminergic-antipsychotic drugs is promptly relieved by parenteral
promethazine, diphenhydramine or hydroxyzine. This is again based on central
anticholinergic action of the drugs
H2 receptor antagonists
Pharmacological actions
1. H2 blockade
• Cimetidine and all other H2 antagonists block histamine-induced gastric secretion, cardiac
stimulation (H2 blockers potentiate histamine induced bronchospasm).
• They attenuate fall in BP due to histamine, especially the late phase response seen with high
doses.
• They are highly selective: have no effect on H1 mediated responses
24.
2. Gastric secretion
•The only significant in vivo action of H2 blockers is marked inhibition of gastric secretion.
All phases (basal, psychic, neurogenic, gastric) of secretion are suppressed dose-
dependently, but the basal nocturnal acid secretion is suppressed more completely.
• Secretory responses to not only histamine but all other stimuli (ACh, gastrin, insulin,
alcohol, food) are attenuated.
• The volume, pepsin content and intrinsic factor secretion are reduced, but the most marked
effect is on acid.
• The usual ulcer healing doses produce 60–70% inhibition of 24 hr acid output. The H2
blockers have antiulcerogenic effect.
• Gastric ulceration due to stress and drugs (NSAIDs, cholinergic, histaminergic) is
prevented.
• They do not have any direct effect on gastric or esophageal motility or on lower
esophageal sphincter (LES) tone.
25.
MOA
Ranitidine
blocks H2 receptorson parietal cells
decreased gastric acid acid secretion
• These drugs bind to the histamine receptors present on the parietal cells and competitively
inhibit the actions of histamine on these receptors and there by reduce gastric acid
secretion
• Both volume and acidity of basal , nocturnal and food induced secretions are reduced
• Inhibition is dose dependent and a single dose can reduce 60-70%gastric secretion for 12
hrs
• Secretion of intrinsic factor , gastrin induced HCl secretion and pepsin are also reduced
26.
Pharmacokinetics
• Rapidly andwell absorbed though bioavailability is 60–80% due to first pass hepatic
metabolism. Absorption is not interfered by presence of food in stomach
• Cimetidine acts for 5-8 hrs , ranitidine and famotidine for 12 hrs
• Partly metabolized in the liver by oxidation and about 2/3rd dose is excreted unchanged in the
urine and bile
• Plasma t1/2 -2-3 hours
• Do not cross BBB due hydrophililicity
Adverse reactions
• H2 blockers are well tolerated (except cimetidine ) with minor side effects like diarrhea ,
dizziness , muscle pain and headache .Because they do not have any significant functions in
other tissues (except stomach)
• On prolonged use of cimetidine ,it may result in gynaecomastia,decreased sperm count,
impotence, galactorrhoea in women
• CNS effects of cimetidine include confusion , restlessness , delirium and hallucination in the
elderly
27.
• Cardiovascular effectslike bradycardia , AV block , cardiac arrest , atrial fibrillation have
been reported with cimetidine and ranitidine
Interactions
1.Cimetidine inhibits the metabolism of many drugs so that they can accumulate to toxic
levels, e.g. theophylline, phenytoin, carbamazepine, phenobarbitone, sulfonylureas,
metronidazole, warfarin, imipramine, lidocaine, nifedipine, quinidine.
2. Antacids reduce absorption of all H2 blockers. When used concurrently a gap of 2 hr
should be allowed.
3. Ketoconazole absorption is decreased by H2 blockers due to reduced gastric acidity.
Therapeutic uses
1. Peptic ulcers –H2 blockers bring about rapid relief from pain and the ulcers heal in 80-
90% of the patients with 6 -8 weeks of treatment
2. Gastritis – H2 blockers are the first line drugs for non-ulcer dyspepsia
28.
3. Zollinger –Ellisonsyndrome-It is a gastric hypersecretory state due to a rare tumour
secreting gastrin. H2 blockers in high doses control hyperacidity and symptoms in many
patients
4. Preanesthetic medication –ranitidine or other H2 blockers may be used to reduce gastric
acid secretion in order to prevent damage to the respiratory mucosa , if aspiration occurs
during surgery
5. GERD-H2 blockers afford symptomatic relief and facilitate healing of esophageal
erosions, but are less effective than PPIs. They are indicated only in mild or stage-1 cases
of GERD
6. Stress induced ulcer -Acutely stressful situations like hepatic coma, severe burns and
trauma, prolonged surgery, prolonged intensive care, renal failure, are associated with gastric
erosions and bleeding.. Intravenous infusion of H2 blockers successfully prevents the gastric
lesions and haemorrhage as well as promotes healing of erosions that have occurred.
29.
5HT antagonists (serotonin antagonists)
• 5HT antagonists or serotonin antagonists are the agents that inhibit the action of serotonin
and serotonergic drugs at serotonin ( 5-HT ) receptors
• These drugs are used for various therapeutic purposes, including the treatment of nausea
and vomiting, migraine headaches, and certain psychiatric disorders.
• There are several subtypes of serotonin receptors (5HT receptors), and drugs within this
class may target specific subtypes
• Here is an overview of the pharmacology of 5HT antagonists
Classification of 5HT Antagonists:
1. 5-HT3 Receptor Antagonists:
Examples: Ondansetron , granisetro , palonosetron , alosetron
2. 5-HT2 Receptor Antagonists:
Examples: ketanserin , cyproheptadine, methysergide, trazodone , nefazodone
30.
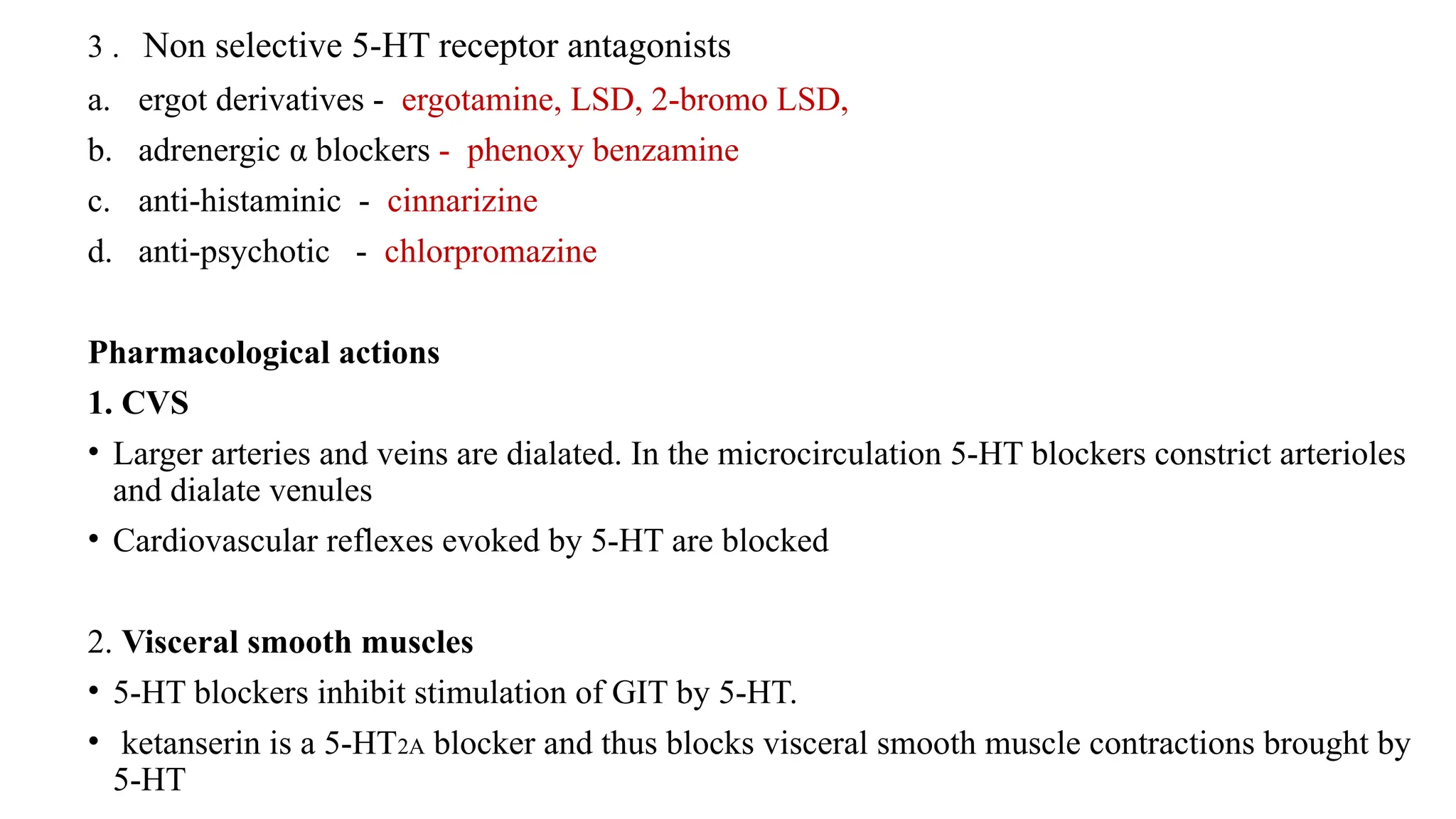
3 . Nonselective 5-HT receptor antagonists
a. ergot derivatives - ergotamine, LSD, 2-bromo LSD,
b. adrenergic α blockers - phenoxy benzamine
c. anti-histaminic - cinnarizine
d. anti-psychotic - chlorpromazine
Pharmacological actions
1. CVS
• Larger arteries and veins are dialated. In the microcirculation 5-HT blockers constrict arterioles
and dialate venules
• Cardiovascular reflexes evoked by 5-HT are blocked
2. Visceral smooth muscles
• 5-HT blockers inhibit stimulation of GIT by 5-HT.
• ketanserin is a 5-HT2A blocker and thus blocks visceral smooth muscle contractions brought by
5-HT
31.
• Ondasetron inhibitsemesis , gut peristalsis by blocking 5- HT3 receptor
3. Nerve endings and adrenal medulla
• Afferent nerve endings activated by 5-HT causing tingling and pricking sensation, as well
as pain are supressed.
• Depolarization of visceral afferents by 5-HT elicits respiratory and cardiovascular reflexes,
nausea and vomiting.These effects are blocked by 5-HT antagonists
• Release of Adr from adrenal medulla in response to 5-HT is abolished
Mechanism of action
5-HT3 Antagonists
Location: Mainly in the CNS and the GI tract, especially the vagus nerve.
Action: Inhibition of 5HT3 receptors reduces the visceral afferent input to the vomiting
center, thus preventing nausea and vomiting.
32.
5-HT2 Receptor Antagonists:
Location:Predominantly in the CNS.
Action: Modulation of serotonin activity, leading to effects such as reduction in migraine
headaches and potential antidepressant or antipsychotic effects.
Pharmacokinetics
Absorption: Varies among different drugs.
Distribution: Extensively distributed in the body, with some crossing the blood-brain
barrier.
Metabolism: Metabolized in the liver, primarily through the cytochrome P450 system.
Excretion: Eliminated through the kidneys.
Adverse reactions
1. 5-HT3 Receptor Antagonists: Headache , Fatigue ,Constipation , light headedness ,
hypotension , bradycardia , QT interval prolongation (particularly with ondansetron)
2. 5-HT2 Receptor Antagonists: Sedation , Anticholinergic effects (dry mouth, blurred
vision), weight gain ,hepatotoxicity
33.
Drug interactions
1. 5-HT3Receptor Antagonists: Potential for drug interactions, especially with medications
that prolong the QT interval.
2. 5-HT2 Receptor Antagonists: Interactions with other drugs affecting serotonin levels in the
CNS
Indications
3. 5-HT3 Receptor Antagonists:
• Prevention and treatment of chemotherapy-induced nausea and vomiting.
• Prevention and treatment of postoperative nausea and vomiting.
• Hyper emesis of pregnancy
• Used in combination with other drugs for the treatment of irritable bowel syndrome (IBS).
2. 5-HT2 Receptor Antagonists:
• Treatment of migraine headaches.
• Adjunctive therapy for certain psychiatric disorders like depression , anxiety
34.
References :
• KDTripathi. Essentials of MEDICAL PHARMACOLOGY, 8th
edition
• Medical pharmacology Padmaja Udaykumar , 7th
edition