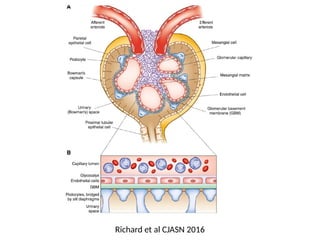
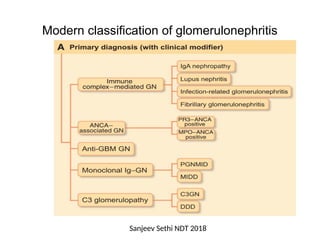
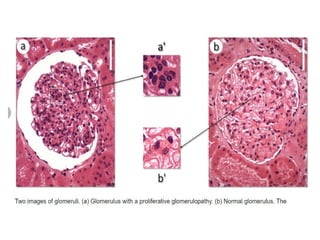
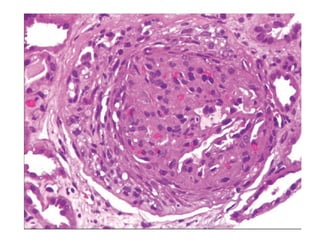
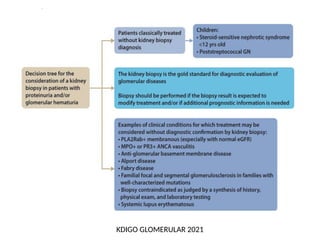
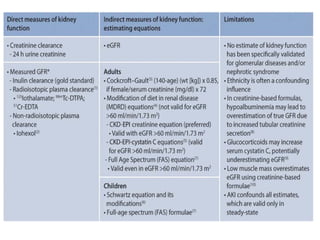
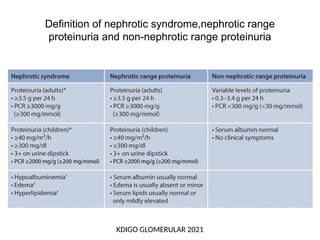
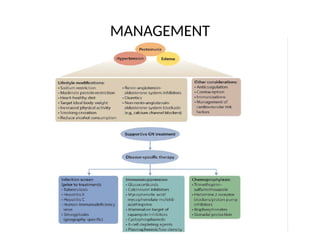
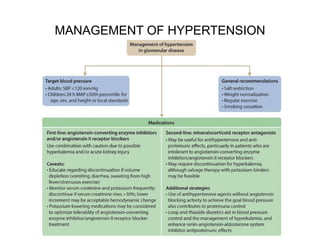
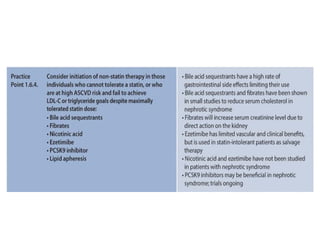
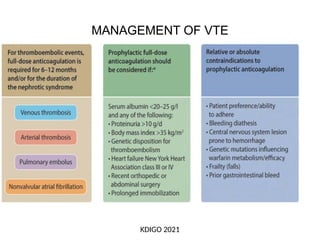
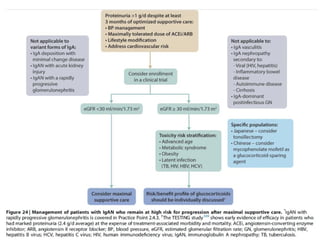
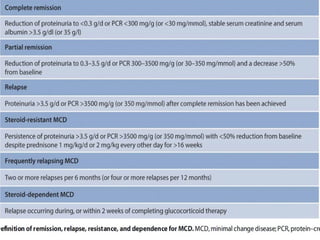
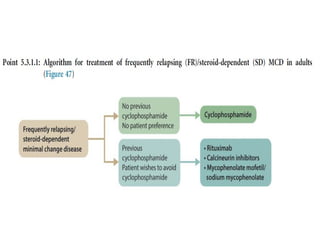
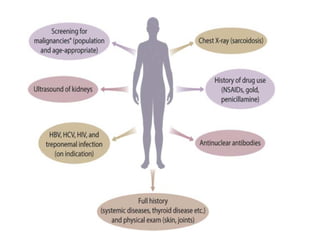
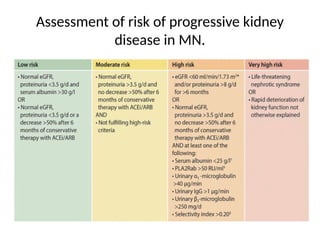
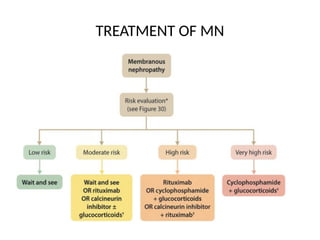
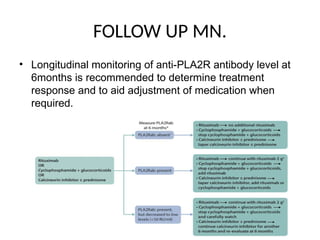
The document provides a comprehensive overview of glomerular diseases, including their anatomy, classification, mechanisms of injury, pathogenesis, and management principles. Key insights include the distinction between primary and secondary glomerular diseases, the clinical presentation of conditions such as IgA nephropathy and minimal change disease, and the importance of kidney biopsy for diagnosis. Management strategies emphasize supportive care, lifestyle modifications, and specific treatments based on the type of glomerular disease.