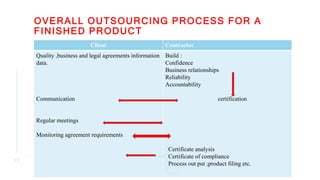
- CROs (Contract Research Organizations) are organizations contracted by pharmaceutical companies to conduct clinical trials and other drug development services. Outsourcing to CROs allows companies to reduce costs and accelerate drug development timelines without requiring large in-house capabilities.
- Key responsibilities of CROs include managing clinical sites and trials, conducting bioanalytical testing, and performing pharmacokinetic analyses. Proper qualification of CROs and ongoing communication are important for successful partnerships. Outsourcing can increase capacity and reduce development cycles for pharmaceutical sponsors.