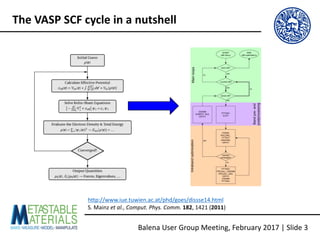
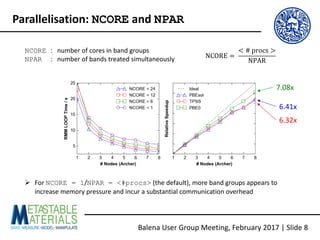
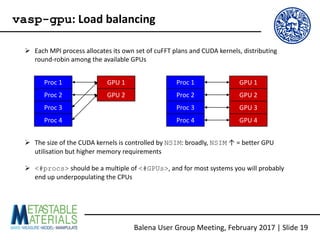
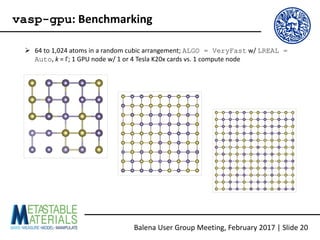
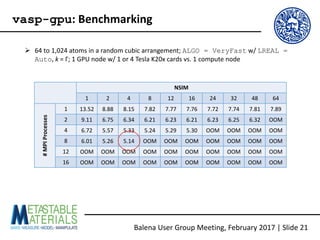
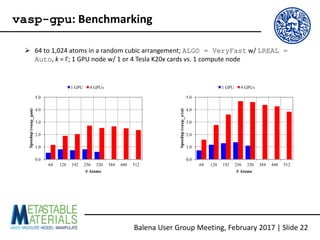
The document discusses the Balena User Group meeting held on February 3, 2017, focusing on the VASP GPU implementation and its parallelization techniques. It highlights the various levels of parallelism in VASP, including k-point and band parallelism, as well as guidelines for optimizing performance on GPU-equipped nodes. Additionally, it covers the compilation and running of VASP-GPU jobs, benchmarking results, and future roadmap points for further improvements.




![MPI processes
KPAR k-point
groups
NPAR band
groups
NGZ FFT
groups (?)
Ø Workload distribution over KPAR k-point groups, NBANDS band groups and NGZ plane-
wave coefficient (FFT) groups [not 100 % sure how this works…]
Balena User Group Meeting, February 2017 | Slide 5
Parallelisation: Workload distribution](https://image.slidesharecdn.com/vasp-gpu-balenausage-somebenchmarks-170203160133/85/vasp-gpu-on-Balena-Usage-and-Some-Benchmarks-5-320.jpg)
![Data
KPAR k-point
groups
NPAR band
groups
NGZ FFT
groups (?)
Ø Data distribution over NBANDS band groups and NGZ plane-wave coefficient (FFT) groups
[also not 100 % sure how this works…]
Balena User Group Meeting, February 2017 | Slide 6
Parallelisation: Data distribution](https://image.slidesharecdn.com/vasp-gpu-balenausage-somebenchmarks-170203160133/85/vasp-gpu-on-Balena-Usage-and-Some-Benchmarks-6-320.jpg)
![Ø During a standard DFT calculation, k-points are independent -> k-point parallelism should
be linearly scaling, although perhaps not in practice:
https://www.nsc.liu.se/~pla/blog/2015/01/12/vasp-how-many-cores
Ø WARNING: <#procs> must be divisible by KPAR, but the parallelisation is via a round-
robin algorithm, so <#k-points> does not need to be divisible by KPAR -> check how
many irreducible k-points you have (IBZKPT file) and set KPAR accordingly
k1
k2
k3
k1 k2
k3
k1 k2 k3
KPAR = 1
t = 3 [OK]
KPAR = 2; t = 2 [Bad]
KPAR = 3
t = 1 [Good]
R1
R2
R3
R1
R2
R1
Balena User Group Meeting, February 2017 | Slide 7
Parallelisation: KPAR](https://image.slidesharecdn.com/vasp-gpu-balenausage-somebenchmarks-170203160133/85/vasp-gpu-on-Balena-Usage-and-Some-Benchmarks-7-320.jpg)





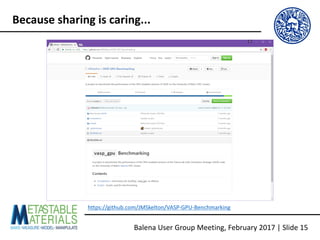

![Ø To use vasp-gpu on Balena, you need to request a GPU-equipped node and perform
some basic setup tasks in your SLURM scripts
#SBATCH --partition=batch-acc
# Node w/ 1 k20x card.
#SBATCH --gres=gpu:1
#SBATCH --constraint=k20x
# Node w/ 4 k20x cards.
##SBATCH --gres=gpu:4
##SBATCH --constraint=k20x
if [ ! -d "/tmp/nvidia-mps" ] ; then
mkdir "/tmp/nvidia-mps"
fi
export CUDA_MPS_PIPE_DIRECTORY=
"/tmp/nvidia-mps"
if [ ! -d "/tmp/nvidia-log" ] ; then
mkdir "/tmp/nvidia-log"
fi
export CUDA_MPS_LOG_DIRECTORY=
"/tmp/nvidia-log"
nvidia-cuda-mps-control -d
https://github.com/JMSkelton/VASP-GPU-Benchmarking/Scripts
Balena User Group Meeting, February 2017 | Slide 17
vasp-gpu: Running jobs](https://image.slidesharecdn.com/vasp-gpu-balenausage-somebenchmarks-170203160133/85/vasp-gpu-on-Balena-Usage-and-Some-Benchmarks-17-320.jpg)












































![ANIMAL_CELL_,_TISSUE_AND_ORGAN_CULTURE[1].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/animalcelltissueandorganculture1-260204172026-4462b440-thumbnail.jpg?width=640&height=640&fit=bounds)






