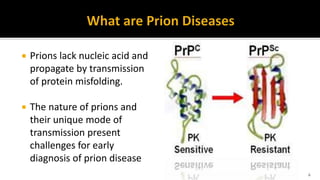
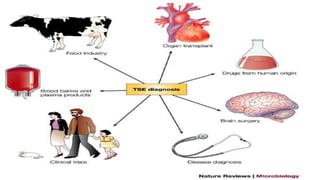
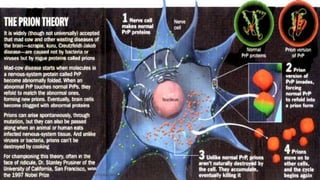
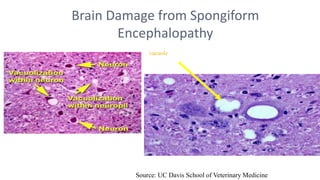
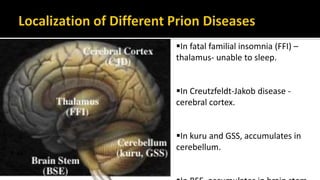
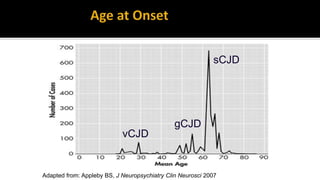
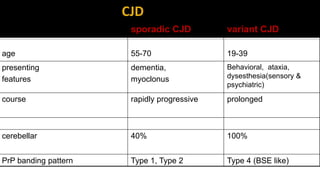
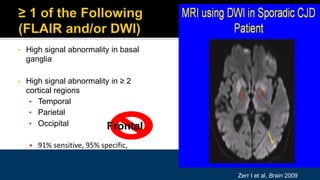
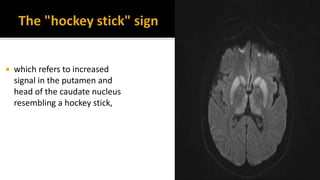
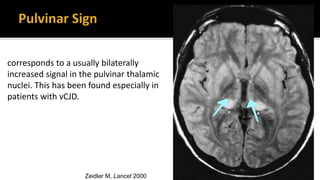
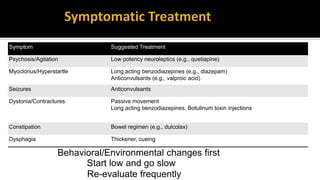
This document discusses prion diseases, also known as transmissible spongiform encephalopathies (TSEs). It notes that TSEs have long incubation periods and progressive clinical courses. They can be caused by conventional or unconventional agents and include diseases like Creutzfeldt-Jakob disease (CJD) in humans and bovine spongiform encephalopathy (BSE) in cattle. Prions, the infectious agents responsible for TSEs, are abnormal forms of proteins that can induce normal proteins to also take the abnormal form. They are resistant to heat, chemicals, and radiation. Diagnosis of prion diseases is difficult due to a lack of immune response or signs of infection. Currently, there




































![ viral encephalitis
Diffuse Lewy body disease
Chronic meningitis
Dementia as a paraneoplastic syndrome
Dementia in motor neuron disease
Limbic encephalitis (and other paraneoplastic syndromes)
Hashimoto encephalopathy (or Steroid-responsive encephalopathy
associated with autoimmune thyroiditis [SREAT])
FTD
ADC](https://image.slidesharecdn.com/prionppt-171015062502/85/Prion-diseases-43-320.jpg)
































































![DUAL AND TRIPLE ANTITHROMBOTIC THERAPY FOR SECONDARY STROKE [Autosaved].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/dualandtripleantithrombotictherapyforsecondarystrokeautosaved-230904113552-c3502b37-thumbnail.jpg?width=640&height=640&fit=bounds)

































