This document summarizes a study that uses coarse-grained molecular dynamics (MD) simulations to model the coordination between competing RNA base pairs. The study uses two RNA systems, each with two separated complementary strands, to test the coarse-grained model. For the first system, the simulation showed little conformational change, but the second system indicated a base pairing interaction as the RMSD sharply decreased partway through the simulation, suggesting the strands bent back together. The coarse-grained model reduces computational expenses compared to all-atom MD while optimizing accuracy and speed for simulating macromolecules like RNA, which could aid drug development efforts.
1. Coordination between competing RNA base pairs using a CG model
Danny Vo, David Bell, and Pengyu Ren*
*University of Texas at Austin
Background
Molecular Dynamics (MD) is a simulation
technique significantly faster than
crystallographic studies, but uses extremely
complex calculations, intensive even for
specialized supercomputers
MD Forces on each atom are calculated, then
their positions are moved using Newton’s laws
of motions
This project focuses on a demonstration of the
coarse grain model which represents
nucleotides with five atoms (as opposed to
thirty), reducing computational expenses while
optimizing accuracy and speed
Being able to simulate macromolecules will aid
drug development, as properties such as binding
sites can be discovered without physical
experiments
Methodology
Tinker: MD engine and Software used to
computationally calculate various parameters
such as Potential Energy, Force Field
Interactions, and Root Mean Square Distance
PyMOL/VMD: Open source imaging system
used to visualize high quality 3D protein
structures and other
molecules/macromolecules
System Setup: Two different RNA systems
were experimented upon. In both systems,
there are two RNA strands with their
complementary base pairs separated. We
then restrained the movement of one of the
strands and executed the simulation
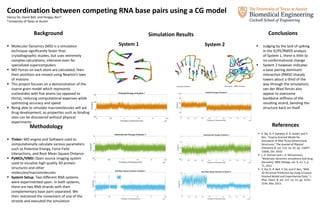
Simulation Results Conclusions
Judging by the lack of spiking
in the IE/PE/RMDS analysis
of System 1, there is little to
no conformational change
System 2 however indicates
a base pairing dominant
interaction (RMSD sharply
lowers about a third of the
way through the simulation).
van der Waal forces also
appear to overcome
backbone stiffness of the
resulting strand, bending the
structure back on itself
References
Z. Xia, D. P. Gardner, R. R. Gutell, and P.
Ren, “Coarse-Grained Model for
Simulation of RNA Three-Dimensional
Structures,” The Journal of Physical
Chemistry B, vol. 114, no. 42, pp. 13497–
13506, Oct. 2010.
J. D. Durrant and J. A. McCammon,
“Molecular dynamics simulations and drug
discovery,” BMC biology, vol. 9, no. 1, p.
71, 2011.
Z. Xia, D. R. Bell, Y. Shi, and P. Ren, “RNA
3D Structure Prediction by Using a Coarse-
Grained Model and Experimental Data,” J.
Phys. Chem. B, vol. 117, no. 11, pp. 3135–
3144, Mar. 2013.
System 1 System 2
Starting orientation Orientation ~7000th timestep