this about the fda regulatory approval process of anda for a generic drug development and the steps which are involved in the regulatory approval of a application and their detailed description and procees
INTRODUCTION
The Abbreviated NewDrug Application (ANDA) Process Is The Regulatory Pathway Managed By
The U.S. Food And Drug Administration (FDA) For The Approval Of Generic Drugs.
It's "Abbreviated" Because Generic Manufacturers Do Not Need To Repeat The Costly And
Lengthy Clinical Trials (Preclinical And Human Studies) To Establish The Drug's Safety And
Effectiveness. Instead, They Must Demonstrate That The Generic Drug Is Therapeutically
Equivalent To An Already Approved Brand-name Drug, Known As The Reference Listed Drug
(RLD).
3.
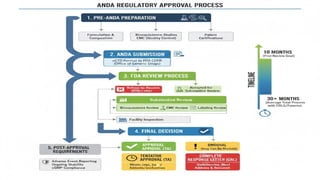
STEPS IN REGULATORYAPPROVAL PROCESS
1. PRE-ANDA PREPARATION
2. ANDA SUBMISSION
3. FDA REVIEW PROCESS
4. FDA DECISION
5. POST-APPROVAL REQUIREMENTS
4.
PRE-ANDA PREPARATION
• PRODUCTSELECTION: Identify The Approved RLD In The FDA's "Orange Book"
(Approved Drug Products With Therapeutic Equivalence Evaluations).
• BIOEQUIVALENCE (BE) STUDIES: Conduct Studies (Usually In Vivo On Healthy
Volunteers) To Scientifically Demonstrate That The Generic Drug Delivers The Same Amount Of
Active Ingredient Into The Bloodstream, At The Same Rate And Extent, As The RLD. This Is
The Core Evidence Of Equivalence.
• CHEMISTRY, MANUFACTURING, AND CONTROLS (CMC): Develop Detailed
Documentation On The Drug's Formulation, Composition, Manufacturing Process, Quality
Control Measures, And Stability Data To Ensure Identity, Strength, Quality, And Purity.
• PATENT CERTIFICATION: The Applicant Must Address Patents Listed In The Orange Book
For The RLD By Filing One Of Four Types Of Certifications (Paragraph I-IV).
5.
PATENT CERTIFICATIONS: ADDRESSINGMARKET
EXCLUSIVITY
Certification Type Generic Applicant States That... Result on Approval
Paragraph I (P-I)
No patent information is listed in the Orange Book for the
RLD.
The ANDA can be approved immediately upon meeting all
other requirements.
Paragraph II (P-II)
The patent(s) listed in the Orange Book have already
expired.
The ANDA can be approved immediately upon meeting all
other requirements.
Paragraph III (P-III)
The patent(s) listed in the Orange Book will expire on a
specific date, and the generic applicant agrees not to market
the drug until that date.
The FDA can give Tentative Approval, with final approval
granted only after the patent expiration date.
Paragraph IV (P-IV)
The listed patent is invalid, unenforceable, or will not be
infringed by the generic drug product. This is a patent
challenge.
This triggers a legal process that may allow for early generic
market entry.
6.
ANDA SUBMISSION
• THEAPPLICANT COMPILES ALL REQUIRED DATA AND DOCUMENTATION INTO AN ELECTRONIC
COMMON TECHNICAL DOCUMENT (ECTD) FORMAT. PAPER SUBMISSIONS ARE NO LONGER
ACCEPTED.
• THE COMPLETE ANDA IS SUBMITTED TO THE FDA'S CENTER FOR DRUG EVALUATION AND
RESEARCH (CDER), OFTEN THROUGH THE ELECTRONIC SUBMISSIONS GATEWAY (ESG).
7.
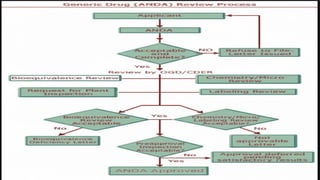
FDA REVIEW PROCESS
•FILING REVIEW: The FDA's Office Of Generic Drugs (OGD) First Reviews The Submission To
Ensure It Is Substantially Complete And Can Be Received For A Full Technical Review. If Incomplete,
The FDA May Issue A Refusal-to-receive (RTR) Letter.
• MULTI-PHASE TECHNICAL REVIEW: The FDA Conducts A Detailed Review Across Multiple
Disciplines:
• BIOEQUIVALENCE: Evaluation Of The BE Study Data.
• CHEMISTRY/MANUFACTURING: Review Of CMC Data And Quality Control.
• LABELING: Assessment Of The Proposed Labeling To Ensure It Is Identical To The RLD's Labeling (With
Certain Permissible Exceptions).
• FACILITY INSPECTIONS: The FDA Will Inspect The Manufacturing, Packaging, And Testing Sites
To Ensure They Comply With Current Good Manufacturing Practice (Cgmp).
9.
FDA DECISION
The FDAWill Conclude The Review With One Of The Following Actions:
• COMPLETE RESPONSE LETTER (CRL): If The FDA Finds Deficiencies (Issues With The
Application, Studies, Or Manufacturing Facilities), A CRL Is Issued. The Applicant Must
Address All Deficiencies And Resubmit An Amended ANDA.
• TENTATIVE APPROVAL (TA): The ANDA Meets All Scientific And Regulatory
Requirements, But The Generic Drug Cannot Be Marketed Yet Due To Unexpired Patents Or
Exclusivities On The RLD. Final Approval Is Granted Once All Legal Barriers Expire.
• FINALAPPROVAL (AP): If All Requirements Are Met, All Facilities Are Deemed Adequate,
And There Are No Blocking Patents Or Exclusivities, The ANDA Is Approved, And The Generic
Drug Can Be Marketed.
10.
POST-APPROVAL REQUIREMENTS
• OnceApproved, The ANDA Holder Must Comply With Ongoing Reporting And Testing
Obligations, Including:
• Periodic Reporting Of Adverse Events.
• Ongoing Stability Testing.
• Adherence To CGMP.
• Submitting Supplements For Changes To The Approved Application.









![REFERENCE
• HTTPS://DOI.ORG/10.1016/J.PHARMA.2025.03.001
• HTTPS://WWW.RESEARCHGATE.NET/PUBLICATION/377784137_REGULATORY_STRA
TEGY_FOR_FILING_
NDAANDA
• AVAILABLEFROM:HTTP://WWW.FDA.GOV/DRUGS/
DEVELOPMENTAPPROVALPROCESS/
HOWDRUGSAREDEVELOPEDANDAPPROVED/APPROVALAPPLICATIONS/
INVESTIGATIONALNEWDRUGINDAPPLICATION/DEFAULT.HTM [CITED 31/3/2021].
• KAUR JASPREET. US FDA DRUG APPROVAL STRATEGIES FOR PHARMACEUTICAL
INDUSTRY. INT J PHARM SCI. MAR–APR 2014:137-46: ARTICLE NO. 24](https://image.slidesharecdn.com/andaregulatroyapprovalprocess-260119034223-3684033f/85/THE-ANDA-REGULATROY-APPROVAL-PROCESS-pptx-12-320.jpg)
















![Abbreviated New Drug Application [ANDA]](https://cdn.slidesharecdn.com/ss_thumbnails/abbreviatednewdrugapplicationanda-160619062810-thumbnail.jpg?width=640&height=640&fit=bounds)






























