Downloaded 28 times








































![МИРОВОЙ ОПЫТ
That each of us is truly biologically unique, extending to even monozy-gotic,
42
“identical” twins, is not fully appreciated. Now that it is possible to
perform a comprehensive “omic” assessment of an individual, including
one’s DNA and RNA sequence and at least some characterization of
one’s proteome, metabolome, microbiome, autoantibodies, and
epigenome, it has become abundantly clear that each of us has truly one-of-
a-kind biological content. Well beyond the allure of the matchless fin-gerprint
or snowflake concept, these singular, individual data and
information set up a remarkable and unprecedented opportunity to im-prove
medical treatment and develop preventive strategies to preserve
health.
FROM DIGITAL TO BIOLOGICAL TO INDIVIDUALIZED MEDICINE
In 2010, Eric Schmidt of Google said “The power of individual targeting —
the technology will be so good it will be very hard for people to watch or
consume something that has not in some sense been tailored for them”
[50]. Although referring to the capability of digital technology, we have now
reached a time of convergence of the digital and biologic domains. It has
been well established that 0 and 1 are interchangeable with A, C, T, and
G in books and Shakespeare sonnets and that DNA may represent the
ultimate data storage system [13, 34]. Biological transistors, also known
as genetic logic gates, have now been developed that make a computer
from a living cell [6]. The convergence of biology and technology was fur-ther
captured by one of the protagonists of the digital era, Steve Jobs,
who said “I think the biggest innovations of the 21st century will be at the
intersection of biology and technology. A new era is beginning” [48].
With whole-genome DNA sequencing and a variety of omic technologies
to define aspects of each individual’s biology at many different levels, we
have indeed embarked on a new era of medicine. The term “personalized
medicine” has been used for many years but has engendered consider-able
confusion. A recent survey indicated that only 4% of the public under-stand
what the term is intended to mean [90], and the hackneyed,
commercial use of “personalized” makes many people think that this
refers to a concierge service of medical care. Whereas “person” refers to
a human being, “personalized” can mean anything from having mono-grammed
stationary or luggage to ascribing personal qualities. Therefore,
it was not surprising that a committee representing the National Academy
of Sciences proposed using the term “precision medicine” as defined by
“tailoring of medical treatment to the individual characteristics of each pa-tient”
[69]. Although the term “precision” denotes the objective of exact-ness,
ironically, it too can be viewed as ambiguous in this context
because it does not capture the sense that the information is derived from
the individual. For example, many laboratory tests could be made more
precise by assay methodology, and treatments could be made more pre-cise
by avoiding side effects — without having anything to do with a spe-cific
individual. Other terms that have been suggested include genomic,
digital, and stratified medicine, but all of these have a similar problem or
appear to be too narrowly focused.
The definition of individual is a single human being, derived from the Latin
word individu, or indivisible. I propose individualized medicine as the pre-ferred
term because it has a useful double entendre. It relates not only to
medicine that is particularized to a human being but also the future impact
of digital technology on individuals driving their health care. There will in-creasingly
be the flow of one’s biologic data and relevant medical informa-tion
directly to the individual. Be it a genome sequence on a tablet or the
results of a biosensor for blood pressure or another physiologic metric
displayed on a smartphone, the digital convergence with biology will de-finitively
anchor the individual as a source of salient data, the conduit of
information flow, and a — if not the — principal driver of medicine in the
future.
THE HUMAN GIS
Perhaps the most commonly used geographic information systems (GIS)
are Google maps, which provide a layered approach to data visualization,
such as viewing a location via satellite overlaid with street names, land-marks,
and real-time traffic data. This GIS exemplifies the concept of
gathering and transforming large bodies of data to provide exquisite tem-poral
and location information. With the multiple virtual views, it gives one
the sense of physically being on site. Although Google has digitized and
thus created a GIS for the Earth, it is now possible to digitize a human
being. As shown in Figure 1, there are multiple layers of data that can
now be obtained for any individual. This includes data from biosensors,
scanners, electronic medical records, social media, and the various omics
that include DNA sequence, transcriptome, proteome, metabolome,
Над решением каких задач работают
лучшие мировые специалисты в
области омиксной науки и на какие
вопросы дают ответы сами эти тех-
нологии? SkАльманах публикует две
лучшие и наиболее убедительные, по
нашему мнению, работы последнего
времени, иллюстрирующие направ-
ление развития научной мысли. Это
статьи “Individualized Medicine from
Prewomb to Tomb” (Cell 157, March 27,
2014) и “Personal Omics Profiling Reve-als
Dynamic Molecular and Medical
Phenotypes” (Cell 148, March 16, 2012),
а также их краткое содержание на
русском языке.
INDIVIDUALIZED MEDICINE FROM
PREWOMB TO TOMB
Eric J. Topol
The Scripps Translational Science Institute, The Scripps Research
Institute and Scripps Health, La Jolla, CA 92037, USA.
Correspondence: etopol@scripps.edu
Reprinted from Cell, 157, Topol E.J., Individualized Medicine from
Prewomb to Tomb, 241-253, 2014, with permission from Elsevier.
http://dx.doi.org/10.1016/j.cell.2014.02.012](https://image.slidesharecdn.com/skbiomed03-10-141201073043-conversion-gate01/85/Sk-biomed-03-10-43-320.jpg)
![Партнер Фонда «Сколково»
43
epigenome, microbiome, and exposome. Going forward, I will use the
term “panoromic” to denote the multiple biologic omic technologies. This
term closely resembles and is adopted from panoramic, which refers to a
wide-angle view or comprehensive representation across multiple appli-cations
and repositories. Or more simply, according to the Merriam-Web-ster
definition of panoramic, it “includes a lot of information and covers
many topics.” Thus the term panoromic may be well suited for portraying
the concept of big biological data.
The first individual who had a human GIS-like construct was Michael Sny-der.
Not only was his whole genome sequenced, he also collected serial
gene expression, autoantibody, proteomic, and metabolomic [11] sam-ples.
A portion of the data deluge that was generated is represented in the
Circos plot of Figure 2 or an adoption of the London Tube map [85]. The
integrated personal omics profiling (iPOP) or “Snyderome,” as it became
known, proved to be useful for connecting viral infections to markedly ele-vated
glucose levels. With this integrated analysis in hand, Michael Sny-der
changed his lifestyle, eventually restoring normal glucose
homeostasis. Since that report in 2012, Snyder and his team have pro-ceeded
to obtain further omic data, including whole-genome DNA methy-lation
data at multiple time points, serial microbiome (gut, urine, nasal,
skin, and tongue) sampling, and the use of biosensors for activity tracking
and heart rhythm. Snyder also discovered that several extended family
members had smoldering, unrecognized glucose intolerance, thereby
changing medical care for multiple individuals.
Of note, to obtain the data and process this first panoromic study, it re-quired
an armada of 40 experienced coauthors and countless hours of
bioinformatics and analytical work. To give context to the digital data bur-den,
it took 1 terabyte (TB) for DNA sequence, 2 TB for the epigenomic
data, 0.7 TB for the transcriptome, and 3 TB for the microbiome. Accord-ingly,
this first human GIS can be considered a remarkable academic feat
and yielded key diagnostic medical information for the individual.
But, it can hardly be considered practical or scalable at this juncture. With
the cost of storing information continuing to drop substantially, the bottle-neck
for scalability will likely be automating the analysis.
On the other hand, each omic technology can readily be undertaken now
and has the potential of providing meaningful medical information for an
individual.
THE OMIC TOOLS
Whole-Genome and Exome Sequencing
Perhaps the greatest technologic achievement in the biomedical domain
has been the extraordinary progress in our ability to sequence a human
genome over the past decade. Far exceeding the pace of Moore’s Law
for the relentless improvement in transistor capacity, there has been a >4
log order (or 0.00007th) reduction in cost of sequencing [10], with a cost
in 2004 of ~$28.8 million compared with the cost as low as $1,000 in
2014 [45]. However, despite this incomparable progress, there are still
major limitations to how rapid, accurate, and complete sequencing can be
accomplished. High-throughput sequencing involves chopping the DNA
into small fragments, which are then amplified by PCR. Currently, it takes
3 to 4 days in our lab to do the sample preparation and sequencing at 303
to 403 coverage of a human genome. The read length of the fragments is
now ~250 base pairs for the most cost-effective sequencing methods, but
this is still suboptimal in determining maternal versus paternal alleles, or
what is known as phasing. Because so much of understanding diseases
involves compound heterozygote mutations, cis-acting sequence variant
combinations, and allele-specific effects, phasing the diploid genome, or
what we have called “diplomics” [93], is quite important. Recently, Mole-culo
introduced a method for synthetically stitching together DNA se-quencing
reads yielding fragments as long as 10,000 base pairs. These
synthetic long reads are well suited for phasing. Unfortunately, the term
“whole-genome sequencing” is far from complete because ~900 genes,
or 3–4% of the genome, are not accessible [65]. These regions are typi-cally
in centromeres or telomeres. Other technical issues that detract from
accuracy include long sequences of repeated bases (homopolymers) and
regions rich in guanine and cytosine. Furthermore, the accuracy for med-ical
grade sequencing still needs to be improved. A missed call rate of 1 in
10,000, which may not seem high, translates into a substantial number of
errors when considering the 6 billion bases in a diploid genome. These er-rors
obfuscate rare but potentially functional variants. Beyond this issue,
the accurate determination of insertions, deletions, and structural variants
is impaired, in part due to the relatively short reads that are typically ob-tained.
The Clinical Sequencing Exploratory Research (CSER) program
at the National Institutes of Health is aimed at improving the accuracy of
sequencing for medical applications [68].
Despite these shortcomings, the ability to identify rare or low-frequency
variants that are pathogenic has been a major outgrowth of high-through-put
sequencing. Well beyond the genome scans and genome-wide asso-ciation
studies that identified common variants associated with most
complex, polygenic diseases and human traits, sequencing leads to high
definition of the uncommon variants that typically have much higher pene-trance.
For example, rare Mendelian conditions have seen a remarkable
surge of definition of their genomic underpinnings [8]. In the first half of
2010, the basis for four rare diseases was published, but in the first half of
2012, that number jumped to 68 [8]. With the power of sequencing, it is
anticipated that the molecular basis for most of the 7,000 known
Mendelian diseases will be unraveled in the next few years.
The 1.5% versus 98.5%
Genome Sequencing Dilemma
The exome consists of only 40 Mb, or 1.5% of the human genome. There
is continued debate over the use of whole exome sequencing compared
with whole-genome sequencing, given the lower cost of sequencing an
exome, that can be readily captured via kits from a few different compa-nies
(Agilent SureSelect, Illumina TruSeq, and Roche NimbleGen).
Exome sequencing is typically performed at much deeper coverage,
>1003 (as compared with 303-403 for whole genome), which enhances
accuracy, and the interpretation of variants that affect coding elements is
far more advanced compared with the rest of the genome. However, the
collective output from genome-wide association studies of complex traits
has indicated that 80% of the incriminated loci are in noncoding regions,
outside the confifnes of genes [56]. It is fair to say that we have long un-derestimated
the importance of the rest of the genome, but its high den-
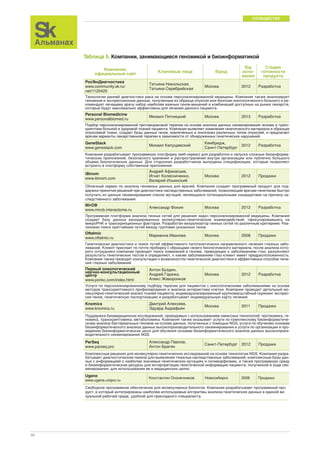
Figure 1. Geographic information system of a human being.
The ability to digitize the medical essence of a human being is predicated
on the integration of multiscale data, akin to a Google map, which consists
of superimposed layers of data such as street, traffic, and satellite views.
For a human being, these layers include demographics and the social
graph, biosensors to capture the individual’s physiome, imaging to depict
the anatomy (often along with physiologic data), and the biology from the
various omics (genome-DNA sequence, transcriptome, proteome, metabo-lome,
microbiome, and epigenome). In addition to all of these layers, there
is one’s important environmental exposure data, known as the “exposome.”](https://image.slidesharecdn.com/skbiomed03-10-141201073043-conversion-gate01/85/Sk-biomed-03-10-44-320.jpg)
![МИРОВОЙ ОПЫТ
sity of key regulatory features provides intricate and extraordinarily tight
control over how genes operate. Recent whole-genome sequencing
studies have identified many critical variants in noncoding portions of the
genome [54]. A typical whole human genome sequence will contain
3.5 million variants compared with the reference genome, predominantly
composed of single-nucleotide polymorphisms but also including inser-tion-
44
deletions, copy number variants, and other types of structural vari-ants
[29]. Today, analysis of most of the 3.5 million variants is left with the
“variant of unknown significant” (VUS) diagnosis. As more people get se-quenced
with the full range of disease phenotypes, the proportion of VUS
will drop, and each sequence will become more informative. Figure 3 pro-vides
a theoretical plot of how further reduction of the cost of whole-genome
sequencing will also be accompanied by large numbers of
individuals undergoing sequencing. In 2014, still well under 100,000 peo-ple
have had whole-genome sequencing with only a very limited number
of phenotypes addressed. At some point in the future, sequence data get
progressively more informative at a lower price point, thus establishing
particular value of whole-genome sequencing. It is not just about getting
a large number of people with diverse medical conditions and diverse an-cestries
sequenced. Here, too much focus on the individual can result in
a loss of context, back to our analogy of the Google map of maximal
zoom obscuring understanding. By anchoring the genomics of family
members, such as was done with the important discovery of PCSK9 rare
variants [40] in cholesterol metabolism, progress in genomic medicine will
be catalyzed.
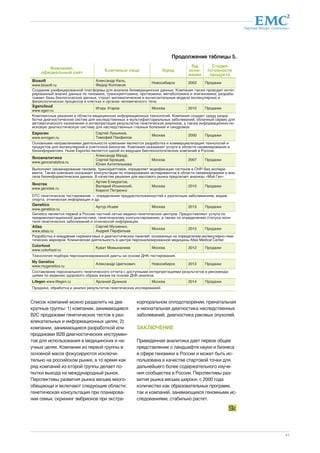
Figure 2. Plots of panoramic information.
Top: Circos plot of the Snyder genome. From outer to inner rings: chromo-some
ideogram; genomic data (pale blue ring), structural variants >50 bp
(deletions [blue tiles], duplications [red tiles]), indels (green triangles); tran-scriptomic
data (yellow ring), expression ratio of viral infection to healthy
states; proteomic data (light purple ring), ratio of protein levels during
human rhinovirus (HRV) infection to healthy states; transcriptomic data (yel-low
ring), differential heteroallelic expression ratio of alternative allele to ref-erence
allele for missense and synonymous variants (purple dots) and
candidate RNA missense and synonymous edits (red triangles, purple dots,
orange triangles, and green dots, respectively). From [11] with permission.
Bottom: Adopted London Tube model of integrated omics from Shendure
and Aiden Integration of the many applications of next-generation DNA se-quencing,
which include sites of DNA methylation (methyl-seq), protein-
DNA interactions (ChIP-seq), 3D genome structure (Hi-C), genetically
targeted purification of polysomal mRNAs (TRAP), the B cell and T cell
repertoires (immunoseq), and functional consequences of genetic variation
(synthetic saturation mutagenesis) with a small set of core techniques, rep-resented
as open circles of “stations.” Like subway lines, individual se-quencing
experiments move from station to station until they ultimately
arrive at a common terminal — DNA sequencing. From [85] with permis-sion.
At this juncture, however, it appears that exome and whole-genome se-quencing
provide complementary information. As the cost of whole-genome
sequencing is further reduced, along with the availability of
enhanced analytical tools for the nongene 98.5% content interpretation,
exome sequencing may ultimately become obsolete.
Single-Cell Sequencing
The ability to perform sequencing of individual cells has provided remark-able
new insights about human biology and disease [4, 71, 83]. The unex-pected
heterogeneity in DNA sequence from one cell to another, such as
has been well documented in tumor tissue and even in somatic cells in
healthy individuals, has enlightened us about intraindividual genomic vari-ation.
The concept of “mosaicism” has gained rapid acceptance — with
multiple mechanisms — ranging from gamete formation, embryonic de-velopment,
to somatic mutation in cells in adulthood, that account for why
each of us has cells with different DNA sequences [62, 63, 73, 97]. It re-mains
unclear whether mosaicism has functional significance beyond
being tied to certain congenital conditions and cancers, but this is an ac-tive
area of research that is capitalizing on single-cell sequencing technol-ogy.
This is especially the case in neuroscience in order to explain the
observed frequent finding of transposons, which appear to involve be-tween
80 and 300 unique insertions for each neuron and are potentially
associated with neurologic diseases [73].
Sperm, which tend to swim solo, are particularly well suited for single-cell
genomics. This work has quantified recombination rates of 25 events per
sperm, identified the hot spots where these events are most likely to
occur, and determined genomic instability as reflected by the rate of de
novo mutations [73, 97]. Such de novo mutations, which increase in
sperm with paternal age, are associated with autism, schizophrenia, and
intellectual disability [8, 57, 73, 94].
Intriguing, and possibly revolutionary, single-cell methods using in situ se-quencing
protocols are set to offer precise spatial information in addition to
linear sequence data. In situ sequencing holds the potential to resolve the
spatial distribution of copy number variants, circular DNA, tumor hetero-geneity,
and RNA localization. A number of methods have been published
in the last year, and progress is likely to accelerate in the near future.
Transcriptomics, Proteomics, and Metabolomics
As opposed to the DNA sequence, which is relatively static, RNA reflects
the dynamic state of the cell. Gene expression of a particular tissue of the
whole genome has been available via microarrays for several years, but
RNA sequencing (RNA-seq) is a relatively new tool that transcends sim-](https://image.slidesharecdn.com/skbiomed03-10-141201073043-conversion-gate01/85/Sk-biomed-03-10-45-320.jpg)
![Партнер Фонда «Сколково»
45
ple expression by capturing data on gene fusions, alternative spliced tran-scripts,
and posttranscriptional changes, along with the whole gamut of
RNAs (including microRNA [e.g., miRNAseq], small RNA, lincRNA, ribo-somal
RNA, and transfer RNA).
A particularly valuable metric related to RNA is the expression quantitative
trait locus (eQTL). By having both genome-wide association study
(GWAS) data and whole-genome gene expression at baseline with or
without particular stimuli, functional genomic assessment has been en-abled.
For example, Westra et al. [99] used eQTLs and loci derived from
GWAS to provide functional genomic, mechanistic insights for multiple
complex traits, including lupus and type1diabetes.
The proteome, metabolome, and autoantibody landscape can be as-sessed
for an individual approaching the whole-genome level via recent
advances in mass spectrometry and protein arrays. Using these tech-niques,
posttranslational modifications of proteins, protein-protein interac-tions,
or the small-molecule metabolites produced by these proteins can
be revealed. Emerging technologies such as RNA-mediated oligonu-cleotide
annealing, selection, and ligation sequencing (RASL-seq), bar-coded
small hairpin RNA (shRNA) libraries, and combinatorial antibody
libraries provide inexpensive and efficient views of biology. Longer read
sequencing provides the opportunity to sequence antibodies, which typi-cally
have variable and constant regions composed of 2,000 nucleotides.
Microbiome
Perhaps no area of biology has received more attention in recent years
than the microbiome. Just the gut microbiome has orders of magnitude
more DNA content than germline human DNA and has markedly height-ened
diversity. Our commensal bacterial flora has been shown to play an
important role in various medical conditions [12]. From fecal samples using
a 16S ribosomal amplicon sequencing method, the gut micro-biome has
been the subject of intensive prospective clinical assessment. It was deter-mined
that there were three major enterotypes of the intestinal microbiome
based on the predominant bacterial species, such as Bacteroides, Ru-minococcus,
or Prevotella [3]. The resident species appear to be quite sta-ble
over an extended period of time and to be initially transmitted via the
mother at childbirth [23]. As the interface between genomics and the host’s
environment, the microbiome clearly plays a pivotal role in defining each
individual. The influence of the diet on the gut microbiome, such as the
content of fiber, along with the underpinning of malnutrition, has been doc-umented
[35, 76]. For example, even an individual’s response to medica-tions,
such as digoxin [39], or multiple drugs used for cancer, has been
shown to be linked to the bacterial flora of the gut microbiome [47, 95].
Epigenome
There has been extraordinary progress in our ability to map the human
epigenome from DNA methylation to histone modifications and chromatin
structure [78, 104]. The prolific ENCODE project has provided troves of
data detailing the role of regulatory elements such as enhancers and insu-lators
and how they are tied to DNA methylation and histone changes [16].
Like gene expression, epigenomic findings are highly cell-type specific,
with more than 200 different cell types in the human body. For methylation,
whole-genome bisulphite sequencing has recently been performed for 30
diverse human cell types [78]. Epigenomic reprogramming has a clear-cut
role in cancer, be it via transcription factors or chromatin regulators [16,
91]. Although access to tissue to define epigenomic signatures is a limiting
factor outside of the cancer space, it is apparent that many other diseases
are affected by epigenomic dynamics, such as complications of diabetes,
rheumatoid arthritis, or hypertension [28, 60, 72]. Furthermore, epigenomic
changes affect susceptibility to diseases, as has been shown with open
chromatin related to the TCF7L2 gene [37] and parental origin of se-quence
variants for Type2 diabetes mellitus and breast and prostate can-cer
[57]. This parent-of-origin issue may be tied to transgenerational
epigenomic instability, as has been well documented in plants, and is cer-tainly
a key element of human biology and heritability [81].
Physiome and Exposome
Understanding and quantifying an individual’s physiology and environ-mental
interactions are crucial to digitizing a human being. Through wear-able
biosensors and smartphones, this has become eminently practical.
Continuous tracking is now obtainable for most key physiologic metrics,
including blood pressure, heart rhythm, glucose, blood oxygen saturation,
brain waves, intraocular eye pressure, and lung function indices. Similarly,
there are environmental sensors that connect with smartphones to quan-tify
such indices as air pollution, pollen count, radiation, water quality, am-bient
humidity, electromagnetic fields, and the presence of pesticides in
food.
Bioinformatics
Fundamental to individualized medicine is the ability to analyze the im-mense
data sets and to extract all of the useful, salient information. This is
exemplified by the task of sifting through a trio of whole-genome se-quences
to find a causative mutation in a proband with an undiagnosed
disease. Typically, this translates to finding one critical nucleotide variant
out of well over one to two million single base variants and simplifying the
analysis by only considering variants that change amino acid sequence or
lead to obvious splicing defects [64]. Identifying the signal from the noise,
with the vast majority of variants categorized as “unknown significance”
(VUS), is the crux of the challenge. Moreover, the tools to assess struc-tural
variants and indels are not as extensively developed and validated.
So there are considerably more data that come from the sequencer for an
individual than can be fully and accurately mined. Beyond this, there is
the need for better integration of the multiple GIS layers, such as
panoromic and biosensor data, and the ability to provide an integrative
multiscale approach to an individual’s data set. Although not the compre-hensive
multilayer as depicted in Figure 2, Zhang et al. [103] recently
used an integrated systems approach, including omics of both human
Figure 3. Hypothetical plot of cost of sequencing and number of individu-als
sequenced over the next 6 years.
As of early 2014, <100,000 individuals have had whole-genome sequencing,
leaving the information difficult to fully interpret (of limited informativeness or
value). When millions of people undergo sequencing, with the full gamut of di-verse
phenotypes and ancestries, and the cost for sequencing continues to
drop, a virtuous cycle of informativeness is established. With the new capabil-ity
in 2014 to have whole-genome sequencing at a cost of $1,000, along with
extremely high throughput, it is likely that millions of individuals will be se-quenced
in the next 3-4 years. The cost of sequencing will continue to drop
throughout this time, as increasing numbers of individuals undergo sequenc-ing.
Projections suggest that at least 20,000 individuals with each phenotype
may be necessary to reliably identify rare, functional genomic variants. Ac-cordingly,
once millions of individuals across all main phenotypes and ances-tries
are sequenced, there is a new set point, or threshold, of informativeness.](https://image.slidesharecdn.com/skbiomed03-10-141201073043-conversion-gate01/85/Sk-biomed-03-10-46-320.jpg)
![МИРОВОЙ ОПЫТ
and mice brains to discover genetic networks in Alzheimer’s disease. Al-though
46
in the past there were generally insufficient efforts to understand
epistasis, gene-gene interactions have been upstaged by the complexity
of a higher-order bioinformatics challenge.
HOW THE OMIC TOOLS REBOOT MEDICINE
A Prewomb-to-Tomb Assessment
At many points in the span of a lifetime, the unique biology of the individ-ual
will play an increasing role. As depicted in the time-line of Figure 4, I
will go through each topic sequentially.
Preconception
The ability to determine carrier mutations for each prospective parent has
been greatly enhanced through multiple direct-to consumer sources, in-cluding
23andMe, Counsyl, GenePeeks, and Good Start Genetics. This
can be considered the ultimate form of prevention for major recessive
conditions and has only received modest attention to date. Counsyl
screens for more than 100 recessive Mendelian traits, and 23andMe
screens 50 carrier conditions. The carrier rates for many serious condi-tions
are higher than most people would suspect, such as 1 in 35 for
spinal muscular atrophy, 1 in 40 for cystic fibrosis, and 1 in 125 individuals
for Fragile X syndrome [92]. Gene-Peeks uses carrier data from 100,000
DNA sequence variants for each prospective parent to perform a com-puter
simulation of 10,000 ‘‘digital babies,’’ determining the probabilistic
odds of significant Mendelian disorders [14]. They are already using their
analytic methodology to screen sperm from the Manhattan cryobank; until
now, sperm banks have been completely unregulated and without ge-nomic
assessment [2, 77]. The concept of higher DNA resolution precon-ception
screening is attractive, given that there are many more
pathogenic variants in the genes that are implicated in disease, such as
cystic fibrosis (2,000 variants), than are conventionally assessed [89].
Fetal Sequencing
Whereas the diagnosis of chromosomal aberrations such as trisomy-21,
18, 13 required amniocentesis or chorionic villi sampling, there are now
four different maternal blood sampling assays to accomplish the same as-sessment
with extremely high (>99%) accuracy [67]. Relying on the
plasma fetal DNA present in adequate quantity from a maternal blood
sample at 8–10 weeks of pregnancy, such testing has been transforma-tive,
preempting the need for amniocentesis in all but the rare exception
when results are ambiguous. Here is a great example of using plasma-free
DNA sequencing to avoid an invasive test that carries a small but im-portant
risk of miscarriage. However, with more than 4 million births in the
United States each year, only a tiny fraction (<2%) of prenatal maternal
blood sampling has yet been performed in clinical practice. Multiple
groups have demonstrated the ability to do a fetal whole-exome se-quence
from a maternal blood sample [24] or whole-genome sequencing
from both parents’ DNA along with the maternal plasma-free DNA [55,
61], but this takes a rather extensive computing and bioinformatics effort
that is not presently scalable. Undoubtedly, that will be resolved over time
but will engender the serious bioethical issues of what constitutes the ap-propriate
reasons for termination of pregnancy. But, at the same time, it
will afford the opportunity to make the molecular diagnoses of conditions
in utero and facilitate treatment then or at the earliest time after birth.
Neonatal Sequencing
Monogenic diseases, many of which present in the first month of life, are
a major cause of neonatal fatality and morbidity [80]. Despite routine heel
sticks for blood sent out for analysis, with attendant delays of several days
to weeks in obtaining results, there has not been any improvement in re-ducing
neonatal mortality related to genetic disorders in the past 20 years
[51, 89]. Now, it has been shown that whole-genome sequencing of new-borns
can be accomplished in <48 hr and can lead to highly actionable in-formation
for managing a neonate’s condition, such as in the classic
example of phenylketonuria or galactosemia, whereby irrevocable dam-age
might otherwise occur [51, 80, 89].
Undiagnosed, Idiopathic, and Rare Diseases
The diagnosis of an XIAP mutation in a child with fulminant pan-colitis with
successful, curative treatment is often cited as the first case of sequencing
to save an individual’s life [101]. Since that report, there have been several
other cases that used whole-genome or exome sequencing, along with
other omic tools, for making the molecular diagnosis of idiopathic condi-tions
[49]. For example, the National Institutes of Health Undiagnosed Dis-
Figure 4. Timeline of Sequencing Applications in
Medicine from Prewomb to Tomb.
The medical application of genomics is relevant to
many points during an individual’s lifespan. Prior to
conception, a couple can have genomic screening
for important recessive alleles. An expectant mother,
at 8–12 weeks of pregnancy, can now have single
tube of blood used to accurately assess chromoso-mal
abnormalities of the fetus, determine gender,
and even have whole-genome sequencing of the
fetus performed. At birth, sequencing the genome of
the newborn can be used to rapidly diagnosis many
critical conditions for which a time delay, which fre-quently
can occur with the present heel stick
screening methods, might lead to irrevocable dam-age.
The molecular basis for serious, undiagnosed
conditions can often be established by sequencing
the individual with parents of siblings. Ultimately,
omic information at a young age will be useful by
providing susceptibility to various medical condi-tions
that have actionable prevention strategies. Se-quencing
can be done to define a pathogen for
more rapid and accurate approaches to infectious
diseases. The driver mutations and key biologic un-derpinning
pathways of an individual’s cancer can
frequently be pinpointed by omics. The root causes
of common polygenic conditions such as diabetes
or coronary heart disease may ultimately be defined
at the individual level. Specific sequence variants of
germline DNA or the gut microbiome have rele-vance
for response to prescription medication.
Defining the genomics of healthspan, rather than
the traditional focus on diseases, may prove to be
especially worthwhile to understand protective alle-les
and modifier genes. For an individual with sud-den
death, a molecular autopsy via sequencing can
be performed, along with family survivors, to deter-mine
the cause of death and potentially prevent un-timely
or avoidable deaths of members of the family
and subsequent generations.](https://image.slidesharecdn.com/skbiomed03-10-141201073043-conversion-gate01/85/Sk-biomed-03-10-47-320.jpg)
![Партнер Фонда «Сколково»
47
ease Program uses exome sequencing to facilitate the diagnosis [30]. Re-cently,
the group at Baylor College of Medicine published a series of 250
individuals, of whom 80% were pediatric and largely affected by neurologic
conditions, who underwent whole-exome sequencing. In that cohort, there
were many affected patients with a known Mendelian trait but without a
specific root cause established. A molecular diagnosis was made in 25%
of the cohort [102]. At the Scripps Health and Scripps Research Institute,
we have screened more than 100 individuals for the potential of having an
idiopathic disease. This requires review by a multidisciplinary physician
panel to assure that a comprehensive evaluation of the patient has been
performed before turning to DNA sequencing. The first of 15 individuals
who we enrolled into our protocol was 16 years old and had an incapaci-tating
neurologic condition. She and her parents were sequenced, and in
Figure 5, the bioinformatics challenge of interpreting the three whole-genome
sequences is presented, along with the molecular diagnosis of an
ADCY5 mutation that had not been previously described. In our 15
probands at Scripps, we have had a successful molecular diagnosis in 8
individuals. However, establishing the diagnosis represents only the first
step of the desired strategy, as providing an effective treatment is the fun-damental
goal. Unfortunately, the number of individuals for whom that has
been achieved is quite limited to date, but strategies using repurposing of
existing drugs, drugs that were partially developed but not commercialized,
or acceleration of the development of genomically guided therapies are all
actively being pursued. With an estimate of at least one million individuals
in the United States with a serious medical condition left without a diagno-sis,
such progress is encouraging [49].
Disease Prevention
At some point in the future, it is hoped that having DNA sequence infor-mation
will pave the way for prevention of an individual’s predisposed
conditions. To date, however, that concept has not been actualized for a
few principal reasons. First, most of the complex, polygenic traits have not
had much more than 10% of their heritability explained by the common
variants assessed by GWAS. The “missing” or unsolved heritability de-tracts
from the ability to assign an individual any certainty or risk or protec-tion
from a particular condition. There are some notable exceptions, such
as age-related macular degeneration or type 1 diabetes mellitus, in which
combinations of common and rare variants can provide a well-character-ized,
quantified risk profile. Second, there is the appropriate question of
whether the knowledge of a risk allele is actionable. Prototypic here is the
apoε4 allele, which carries an unequivocal high risk for Alzheimer’s dis-ease,
yet there is no proven strategy to prevent the disease. So, even
armed with known risk, there is a lack of knowledge for how to mitigate it.
Third, the way data for genomic susceptibility are analyzed via a popula-tion
approach makes it difficult to extrapolate such average findings to a
particular individual. For example, someone may have low-frequency
modifier genes for a condition at risk or unusual environmental interac-tions
that markedly affect susceptibility. Notwithstanding these issues, as
millions of individuals with diverse phenotypes undergo whole-genome
sequencing (Figure 3), the ability to provide meaningful risk data will in-crease.
Having the full GIS of each individual will further enrich the proba-bilistic
approach of providing vulnerability data early in one’s life. For
example, if one’s sequence data indicate a risk for hypertension, that risk
may be further modulated by knowledge of his/her proteins, metabolites,
microbiome, and epigenomics. Specific treatments could be used that are
biologically based from one’s GIS. Similarly, for asthma, the panoromic in-formation,
coupled with biosensors that track air quality, pollution, forced
expiratory volume, and other relevant physiologic metrics, could prove
useful to prevent an attack.
A futuristic way in which genomics and biosensors will ultimately converge
is through injectable nanosensors that put the blood in continuous surveil-lance
mode [25]. Such sensors have the ability to detect a DNA, RNA,
autoantibody, or protein signal and to wirelessly transmit the signal to the
individual’s smartphone. This sets up the potential for detecting endothe-lial
sloughing from an artery before a heart attack [15], plasma tumor DNA
in a patient being treated for or in remission from cancer, or a child with
known genomic risk of autoimmune diabetes that is developing autoanti-bodies
to pancreatic b-islet cells long before there has been destruction.
The blood under continuous surveillance concept highlights the potential
ability to temporally detect a risk signal for a major clinical event. That
could be intensive antiplatelet medication to prevent a heart attack, ge-nomic-
guided treatment of cancer recurrence at the earliest possible junc-ture,
or immunomodulation therapy for autoimmune diabetes.
Infectious Diseases
Whole-genome sequencing has proven to be particularly useful for track-ing
pathogen outbreaks, such as for tuberculosis [32], methicillin-resistant
Staphylococcus aureus, antibiotic-resistant Klebsiella pneumonia, and
Clostridium difficile [22, 44, 87]. Beyond identifying the particular bacterial
or viral strain that accounts for an outbreak’s origin and spread, sequenc-ing
is likely to prove to be quite useful for rapid, early characterization of
the cause of infection and specific, effective antibiotic therapy. For sepsis,
the current standard of care is to take blood or other body fluids for cul-ture,
which typically takes 2 days to grow out. Additionally, there is at least
another day required to determine sensitivities to a range of antibiotics. In
the future, with lab-on-a-chip sequencing platforms that attach to or are in-tegrated
with a smartphone (Biomeme and QuantuMD), it may be feasi-ble
to do rapid sequencing of the pathogen and determination of the
optimal treatment. Such a strategy would preempt the need for broad-spectrum
antibiotic use, and the rapid diagnosis and targeted treatment
would likely have a favorable impact on prognosis in these very high-risk,
critically ill patients.
Figure 5. Legend from Whole-Genome Sequencing to Identification of a
Causative Variant.
As of early 2014, <100,000 individuals have had whole-genome sequencing,
leaving the information difficult to fully interpret (of limited informativeness or
value). When millions of people undergo sequencing, with the full gamut of di-verse
phenotypes and ancestries, and the cost for sequencing continues to
drop, a virtuous cycle of informativeness is established. With the new capabil-ity
in 2014 to have whole-genome sequencing at a cost of $1,000, along with
extremely high throughput, it is likely that millions of individuals will be se-quenced
in the next 3-4 years. The cost of sequencing will continue to drop
throughout this time, as increasing numbers of individuals undergo sequenc-ing.
Projections suggest that at least 20,000 individuals with each phenotype
may be necessary to reliably identify rare, functional genomic variants. Ac-cordingly,
once millions of individuals across all main phenotypes and ances-tries
are sequenced, there is a new set point, or threshold, of informativeness.](https://image.slidesharecdn.com/skbiomed03-10-141201073043-conversion-gate01/85/Sk-biomed-03-10-48-320.jpg)
![МИРОВОЙ ОПЫТ
Cancer
With cancer’s basis in genomics, there have been extensive efforts to
characterize the principal driver mutations and biologic pathways, espe-cially
48
through The Cancer Genome Atlas (TCGA) [1, 52, 53]. Our under-standing
of the biology of cancer has expanded exponentially and, with it,
so has the appreciation for its extreme complexity. Perhaps the two clas-sical
“Hallmarks of Cancer” Reviews in Cell, one in 2000 and the sequel
in 2011, best exemplify this [41, 42]. The diagram to explain the principal
mechanisms of cancer was already exceptionally complex in 2000 and
became at least a log order more intricate a decade later. In a more re-cent
review of the cancer genome landscape, the Johns Hopkins group
provided perspective for the 84 known oncogenes and 54 tumor suppres-sor
genes that have been fully validated [96]. There will unquestionably
be more, but estimates of the total number of genes involved in pivotal
mutations may wind up being 200. Beyond this, the principal pathways in-volving
cell survival, cell fate, and DNA damage repair are recognized.
Certain cancers have a relative low burden of mutations per megabase of
the tumor genome, such as acute myelogenous leukemia (<1), whereas
other are quite high, like lung adenocarcinoma or squamous cell carci-noma
(50) [53, 96]. Mutations of certain genes, such as the P53 tumor
suppressor, are found in some patients with any of 12 common forms of
cancer [53]. Our old taxonomy of cancer based upon the organ of origin
may be considered inapt, for knowledge of the driver mutation(s) and
pathway could be more useful for individualizing treatment. “Nof1” case
reports with whole-genome sequencing have been particularly illuminat-ing
for the clonal origin of an individual’s cancer [9, 38]. In the past 2
years, the FDA has approved almost 20 new drugs that target a specific
mutation for cancer.
So, with these leaps in understanding biology and introduction of new ther-apies,
why has there been relatively little impact in the clinic to date? One
major barrier is that we do not have drugs that can target tumor suppres-sor
genes, making up 40% of mix of principal, driver mutations. Some-times
there are workarounds for this issue, such as the tumor suppressor
gene PTEN, which results in PI3-kinase activation, but more often, this is
not the case. Even for oncogenes, less than 40% have a specific drug an-tagonist,
as most are part of protein complexes [96]. A second critical issue
is that there is marked heterogeneity in tumors, both within an individual’s
primary tumor and certainly intermetastatic. This appears to be a founda-tion
for the common occurrence of relapse after an initial marked re-sponse,
reflecting success directed to an oncogene but also that other
undetected mutations become capable of propagating the tumor. The
BRAF mutations, which are drivers in a variety of tumors, notably
melanoma, thyroid, and colon, can be treated with a specific BRAF in-hibitor.
In the first 2 weeks of oral therapy, there is usually a marked re-sponse,
but at 9-12 months, a relapse is quite typical [88]. Interestingly,
when targeting BRAF for colon cancer, there appears to be primary resist-ance
to these inhibitors [74] related to EGFR expression and emphasizing
that the stroma, microenvironment of the tumor can still exert an important
role. The issues of heterogeneity and resistance lend credence to the use
of combinations of targeted drugs in the future, but that has yet to be ex-plored
at scale in prospective trials. A third largely unaddressed issue in
the clinic is the involvement of the epigenome in tumorigenesis. At least 40
epigenome regulator genes are known that have highly recurrent somatic
mutations in tumors across a variety of cancers, affecting multiple target
genes simultaneously [32, 53, 84, 96]. These are not screened for clini-cally,
nor are there drugs available to modulate their effect.
In the clinic today, the bare bones of mutation screening are typically used,
such as HER2 for breast cancer or KRAS for colon cancer. Recently,
Foundation Medicine commercialized a targeted gene panel of 287 genes
that have an established role in cancer [27]. Using predominantly fixed-for-malin,
paraffin-embedded samples, mutation cell concordance was estab-lished
compared with mass spectrometric methodology (Sequenom), and
the typical driver mutations were identified in a cohort of more than 2,000
individuals, such as TP53, KRAS, CDKN2A, and PIK3CA [27]. However,
there was a long tail of uncommon mutations that was identified, reflecting
the profound diversity of cancers. This panel represents a step forward
compared with a very limited gene mutation screen for commonly occur-ring
drivers, which might even miss other pathogenic mutations within in-criminated
genes. The 287 genes assessed represent only <15% of genes
and only the coding elements. This is in contrast to research studies of
whole-genome and whole-exome sequencing, with paired germline DNA
for each individual, to more precisely determine driver mutations [53]. Fur-ther,
multiple recent studies have highlighted the role of noncoding ele-ments
of the genome to play a prominent role, such as TERT promoters in
melanoma [46], a long, noncoding RNA SChLAP1 for aggressive prostate
cancer [75], and identification of 100 non-coding driver variants for cancer
using a new bioinformatics tool known as FunSeq [54]. Clearly, even a
comprehensive exome would only represent a limited swath of sampling
for root causes of cancer in an individual.
Cancer genomic medicine of the future will likely involve a GIS of the
tumor with assessment of DNA sequence, gene expression, RNA-seq,
microRNAs, proteins, copy number variations, and DNA methylation
cross-referenced with the individual’s germline DNA. But the issue of ad-dressing
heterogeneity still looms [5, 96], and for that, there are a few
possible steps, including deep sequencing of the tumor at multiple loca-tions,
single-cell sequencing, or the use of the “liquid biopsy” of cancer
[26, 59, 82].
Cell-free tumor DNA in plasma, which is present in the vast majority of pa-tients
with cancer, has been shown to be a useful biomarker for following
patients [26, 59, 82] and appears to have independent prognostic signifi-cance
[17]. It may be that plasma tumor DNA is the best representative of
the cancer for targeted treatment because avoidance of metastasis is of
utmost concern. The ease of isolating and sequencing cell-free tumor
DNA is likely to make this a very attractive and routine in the future. Also
of particular interest, at some point, will be screening healthy people for
cell-free tumor DNA to determine whether we are constantly facing micro-scopic
tumor burden but are able to effectively keep the disease in check
by a variety of homeostatic mechanisms.
Molecular Diagnosis
When a patient receives a diagnosis of a chronic illness today, it is non-specific
and is based on clinical and not molecular features. Take, for ex-ample,
diabetes mellitus type 2, which could reflect anything from insulin
resistance, failure of β-islet cells, or a variety of subtypes, including α-
adrenergic receptor (ADRA2A) diabetes [36] or a zinc transporter subtype
(SLC30A8) [86]. Common genomic variants have been identified in path-ways
involving signal transduction, cell proliferation, glucose sensing, and
circadian rhythm [20]. Some individuals with a high fasting glucose have a
G6PC2 variant that is associated with protection from diabetes [7].
Agenotype score, amalgamating the number of risk variants, has been
shown to be helpful for identifying high susceptibility [66]. Moreover, there
are 13 classes of drugs to treat diabetes, and the treatment could be
made considerably more rational with knowledge of the individual’s un-derlying
mechanism(s). This brief summary of the diabetes example re-flects
the need for a new molecular taxonomy across all diseases. When
an individual is diagnosed (or at some point when risk can be defined),
the molecular basis will be assessed and, ideally, when possible, the root
cause will be established. Clearly, for many common diseases, there are
multiple pathways implicated, and this may prove to be difficult. But there
has yet to be a systematic attempt of providing such a molecular diagno-sis
in clinical care. Despite multiple reports of molecular subtypes of
asthma [98], multiple sclerosis [70], and colon [79] and uterine cancer
[52], which appear to be linked with therapy and prognosis, this has yet to
be made part of medical practice.
Pharmacogenomics
Just as molecular subtyping of chronic disease is not part of medical prac-tice,
pharmacogenomics screening for either assurance of efficacy or
avoidance of major side effects is predominantly ignored. With the use of
GWAS, there has been an avalanche of discovery of alleles that are piv-](https://image.slidesharecdn.com/skbiomed03-10-141201073043-conversion-gate01/85/Sk-biomed-03-10-49-320.jpg)
![Партнер Фонда «Сколково»
49
otal to individual drug response. Unlike polygenic disease, for which the
penetrance for a common sequence variant is quite low (approximate
odds ratio of 1.15), the typical genotype odds ratio for prescription drugs
can be as high as 80, and for many, the range is 3- to 40-fold (reviewed in
depth in [43]). The likely explanation for this pronounced impact of com-mon
variants on individual drug response is based on selection — as
compared with diseases, the human genome has had very limited time to
adapt to medication exposure. Despite there being more than 100 drugs
that carry a genomic “label” by the FDA, meaning that there is a recom-mendation
for genotype assessment before the drug is used, there is
rarely any pharmacogenomic assessment in clinical practice. This needs
to improve, and perhaps the availability of point-of-care testing will help,
along with reduced cost, to eventually promote routine use. Beyond this
barrier, there needs to be more genomic sequencing for commonly used
drugs, with associated phenotypic determination of efficacy and side ef-fects,
along with systematic omic assessment for drugs in development.
From the marked success of discovering genomic-drug interactions found
to date, there is certainly the sense that, the more you look, the more you
will find. The potential here is to reduce the waste of pharmaceuticals, not
just by avoiding drugs that will not provide efficacy for particular individu-als
but also by avoiding serious toxicity that can be either fatal or life
threatening.
Healthspan
The human reference genome is based upon multiple young individuals
who had no phenotypic characterization. Accordingly, we know nothing
about the reference human’s natural history of disease, and one can con-sider
this as a flawed standard for comparison. Ideally, we should have a
reference genome that has had rigorous phenotyping. This is especially
the case in an era of using sequencing in medical practice but with an in-adequate
comparator. Perhaps the optimal phenotype would be
healthspan. At Scripps, we have defined healthy elderly as age >80 years
with no history of chronic illness or use of medications. The cohort (known
as “Wellderly”) that we have assembled over the past 7 years of 1,400 in-dividuals
has an average age of 88, and we have completed whole-genome
sequencing for 500 of these individuals. The intent is to provide a
more useful reference genome with a clearly defined, uniform, and rele-vant
phenotype.
Moreover, there is another important application of healthspan genomics.
Multiple studies have established that a research investment in under-standing
healthy aging would be more prudent than in any specific dis-ease
category [33]. Because the cohort that we have enrolled carries a
similar burden of common risk variants for chronic diseases compared
with the general population, there are most likely a substantial number of
modifier genes and protective alleles that may be ultimately identified.
One example is from APP, a gene that has a variant for both high risk of
Alzheimer’s and another rare variant with marked protection from cogni-tive
impairment of Alzheimer’s [19]. Unquestionably, there are many more
such variants left to be discovered, and therein lies the potential for drug
discovery efforts that can follow such findings from nature of particular
genes and pathways that prevent diseases.
Molecular Autopsies
Although physical autopsies have lost favor and become exceptionally
rare, there is an opportunity to use sequencing to determine the cause of
death, particularly when this occurs suddenly. Targeted or whole-genome
sequencing for heritable heart disorders implicated in sudden death, in-cluding
ion channel mutations and hypertrophic cardiomyopathy, can be
performed in the deceased individual and family members. This approach
is now actively being pursued in New York City for all sudden cardiac
deaths [21] and may prove helpful in preventing this condition in family
members.
Future Directions
This prewomb-to-tomb review has emphasized that there is a dispropor-tionate
relationship between knowledge and implementation into clinical
practice. For individualized medicine to take hold, it will require intensive,
rigorous validation that these new approaches improve patient outcomes
and are demonstrated to be cost effective. This proof will be essential for
the medical community to embrace the opportunities but will also require
educational programs that squarely address the knowledge chasm that
currently exists for practicing physicians. A second theme is that our ef-forts
have been largely sequence centric and have not adequately taken
into account or integrated the data from other omics, no less biosensors
and imaging. Related to this deficiency, there is a profound shortage of
data scientists in biomedicine, with unparalleled opportunities to process
enormous, high-yield data sets. While we will increasingly rely on algo-rithms,
artificial intelligence, and machine learning, the rate-limiting step
necessitates talented biocomputing and bioinformatic human expertise.
One of the most attractive outgrowths of defining each individual’s unique
biology in an era with unprecedented digital infrastructure is to be able to
share the data. By taking the deidentified data from each individual, in-cluding
panoromic, biosensors, social graph, treatment, and outcomes,
an extraordinary resource can now be developed. Such a massive open
online medical information (MOOM) repository could provide matching
capability to approximate a newly diagnosed individual’s data as com-pared
with all of those previously amalgamated. For a patient with cancer,
for example, this could provide closest matches to the tumor GIF, demo-graphics,
treatment, and outcomes to select an optimal strategy; this
would potentially take Bayesian principles to a new, enriched potential.
Such a MOOM resource does not need to be confined to cancer, but the
first to be announced was with the Leukemia & Lymphoma Society and
Oregon Health Sciences University for 900 patients with liquid tumors
[100]. Hopefully, this will be one of many data-sharing initiatives in medi-cine
to go forward, now that such rich unique information can be captured
at the individual level, and our computing infrastructure is so well suited to
perform such functions. Although we are still at the nascent stages of indi-vidualized
medicine, there has never been more promise and opportunity
to reboot the way health care can be rendered. Only with systematic vali-dation
of these approaches at the intersection of biology and digital tech-nology
can we actualize this more precise, futuristic version of medicine.
ACKNOWLEDGMENTS
I want to express my gratitude to my colleagues Ali Torkamani, PhD, and
Erick Scott, MD, who reviewed the manuscript and offered editorial input
and to Katrina Schreiber, who helped prepare the typescript and graphics.
This work was supported by NIH/NCATS1UL1TR001114. Dr. Topol is an
advisor to Illumina, Genapsys, and EdicoGenomics and is a cofounder of
CypherGenomics.
Эрик Топол
Индивидуальная медицина: от планирования семьи до смерти
Не все осознают, насколько биологически уникален каждый
человек, даже однояйцевые близнецы. Степень этой индиви-
дуальности стала ясна лишь сейчас, благодаря персонализи-
рованному омиксному профилированию, в первую очередь
ДНК- и РНК-секвенированию и описанию (в большей или
меньшей степени) протеома, метаболома, эпигенома, микро-
биома и профиля антител. Доступность этой информации
создала беспрецедентные возможности для улучшения ме-
дицинской технологии и развития превентивных стратегий
сохранения здоровья. Масштаб этих новых возможностей
значительно шире, чем у уникального генотипирования, с ко-
торым обычно ассоциируют персональную геномику.](https://image.slidesharecdn.com/skbiomed03-10-141201073043-conversion-gate01/85/Sk-biomed-03-10-50-320.jpg)


![МИРОВОЙ ОПЫТ
PERSONAL OMICS PROFILING
REVEALS DYNAMIC MOLECULAR
AND MEDICAL PHENOTYPES
SUMMARY
Personalized medicine is expected to benefit from combining genomic in-formation
52
with regular monitoring of physiological states by multiple high-throughput
methods. Here, we present an integrative personal omics
profile (iPOP), an analysis that combines genomic, transcriptomic, pro-teomic,
metabolomic, and autoantibody profiles from a single individual
over a 14 month period. Our iPOP analysis revealed various medical
risks, including type 2 diabetes. It also uncovered extensive, dynamic
changes in diverse molecular components and biological pathways
across healthy and diseased conditions. Extremely high-coverage ge-nomic
and transcriptomic data, which provide the basis of our iPOP, re-vealed
extensive heteroallelic changes during healthy and diseased
states and an unexpected RNA editing mechanism. This study demon-strates
that longitudinal iPOP can be used to interpret healthy and dis-eased
states by connecting genomic information with additional dynamic
omics activity.
INTRODUCTION
Personalized medicine aims to assess medical risks, monitor, diagnose
and treat patients according to their specific genetic composition and mo-lecular
phenotype. The advent of genome sequencing and the analysis of
physiological states has proven to be powerful [8]. However, its imple-mentation
for the analysis of otherwise healthy individuals for estimation
of disease risk and medical interpretation is less clear. Much of the
genome is difficult to interpret and many complex diseases, such as dia-betes,
neurological disorders and cancer, likely involve a large number of
different genes and biological pathways [6, 17, 33], as well as environ-mental
contributors that can be difficult to assess. As such, the combina-tion
of genomic information along with a detailed molecular analysis of
samples will be important for predicting, diagnosing and treating diseases
as well as for understanding the onset, progression, and prevalence of
disease states [45].
Presently, healthy and diseased states are typically followed using a lim-ited
number of assays that analyze a small number of markers of distinct
types. With the advancement of many new technologies, it is now possi-ble
to analyze upward of 105 molecular constituents. For example, DNA
microarrays have allowed the subcategorization of lymphomas and
gliomas [38], and RNA sequencing (RNA-Seq) has identified breast can-cer
transcript isoforms [29, 33, 52, 56]. Although transcriptome and RNA
splicing profiling are powerful and convenient, they provide a partial por-trait
of an organism’s physiological state. Transcriptomic data, when com-bined
with genomic, proteomic, and metabolomic data are expected to
provide a much deeper understanding of normal and diseased states
[46]. To date, comprehensive integrative omics profiles have been limited
and have not been applied to the analysis of generally healthy individuals.
To obtain a better understanding of: (1) how to generate an integrative
personal omics profile (iPOP) and examine as many biological compo-nents
as possible, (2) how these components change during healthy and
diseased states, and (3) how this information can be combined with ge-nomic
information to estimate disease risk and gain new insights into dis-eased
states, we performed extensive omics profiling of blood
components from a generally healthy individual over a 14 month period
(24 months total when including time points with other molecular analy-ses).
We determined the whole-genome sequence (WGS) of the subject,
and together with transcriptomic, proteomic, metabolomic, and autoanti-body
profiles, used this information to generate an iPOP. We analyzed the
iPOP of the individual over the course of healthy states and two viral in-fections
(Figure 1A). Our results indicate that disease risk can be esti-mated
by a whole-genome sequence and by regularly monitoring health
states with iPOP disease onset may also be observed. The wealth of in-formation
provided by detailed longitudinal iPOP revealed unexpected
molecular complexity, which exhibited dynamic changes during healthy
and diseased states, and provided insight into multiple biological
processes. Detailed omics profiling coupled with genome sequencing can
provide molecular and physiological information of medical significance.
This approach can be generalized for personalized health monitoring and
medicine.
RESULTS
Overview of Personal Omics Profiling
Our overall iPOP strategy was to: (1) determine the genome sequence at
high accuracy and evaluate disease risks, (2) monitor omics components
over time and integrate the relevant omics information to assess the vari-ation
of physiological states, and (3) examine in detail the expression of
personal variants at the level of RNA and protein to study molecular com-plexity
and dynamic changes in diseased states.
We performed iPOP on blood components (peripheral blood mononu-clear
cells [PBMCs], plasma and sera that are highly accessible) from a
Rui Chen,1,11 George I. Mias,1,11 Jennifer Li-Pook-Than,1,11 Lihua
Jiang,1,11 Hugo Y.K. Lam,1,12 Rong Chen,2,12 Elana Miriami,1 Konrad J.
Karczewski,1 Manoj Hariharan,1 Frederick E. Dewey,3Yong Cheng,1
Michael J. Clark,1 Hogune Im,1 Lukas Habegger,6,7 Suganthi Balasub-ramanian,
6,7 Maeve O’Huallachain,1 Joel T. Dudley,2 Sara Hillenmeyer,1
Rajini Haraksingh,1 Donald Sharon,1 Ghia Euskirchen,1 Phil Lacroute,1
Keith Bettinger,1 Alan P. Boyle,1 Maya Kasowski,1 Fabian Grubert,1
Scott Seki,1 Marco Garcia,1 Michelle Whirl-Carrillo,1 Mercedes Gal-lardo,
9,10 Maria A. Blasco,9 Peter L. Greenberg,4 Phyllis Snyder,1Teri E.
Klein,1 Russ B. Altman,1,5 Atul J. Butte,2 Euan A. Ashley,3 Mark Ger-stein,
6,7,8 Kari C. Nadeau,2 Hua Tang,1 and Michael Snyder1
1Department of Genetics, Stanford University School of Medicine; 2Divi-sion
of Systems Medicine and Division of Immunology and Allergy, De-partment
of Pediatrics; 3Center for Inherited Cardiovascular Disease,
Division of Cardiovascular Medicine; 4Division of Hematology, Depart-ment
of Medicine; 5Department of Bioengineering Stanford University,
Stanford, CA 94305, USA; 6Program in Computational Biology and Bio-informatics;
7Department of Molecular Biophysics and Biochemistry;
8Department of Computer Science Yale University, New Haven, CT
06520, USA; 9Telomeres and Telomerase Group, Molecular Oncology
Program, Spanish National Cancer Centre (CNIO), Madrid E-28029,
Spain; 10Life Length, Madrid E-28003, Spain; 11These authors contribu-ted
equally to this work; 12Present address: Personalis, Palo Alto, CA
94301, USA. *Correspondence: mpsnyder@stanford.edu
Reprinted from Cell, 148, Chen R. et al., Personal Omics Profiling Re-veals
Dynamic Molecular and Medical Phenotypes, 1293-1307, 2012,
with permission from Elsevier. ttp://dx.doi.org/10.1016/j.cell.2012.02.009](https://image.slidesharecdn.com/skbiomed03-10-141201073043-conversion-gate01/85/Sk-biomed-03-10-53-320.jpg)
![Партнер Фонда «Сколково»
53
54-year-old male volunteer over the course of 14 months (IRB-8629). The
samples used for iPOP were taken over an interval of 401 days (days 0–
400). In addition, a complete medical exam plus laboratory and additional
tests were performed before the study officially launched (day 123) and
blood glucose was sampled multiple times after the comprehensive omics
profiling (days 401–602) (Figure 1A). Extensive sampling was performed
during two viral infections that occurred during this period: a human rhi-novirus
(HRV) infection beginning on day 0 and a respiratory syncytial
virus (RSV) infection starting on day 289. A total of 20 time points were ex-tensively
analyzed and a summary of the time course is indicated in Figure
1A. The different types of analyses performed are summarized in Figures
1B and 1C. These analyses, performed on PBMCs and/or serum compo-nents,
included WGS, complete transcriptome analysis (providing informa-tion
about the abundance of alternative spliced isoforms, heteroallelic
expression, and RNA edits, as well as expression of miRNAs at selected
time points), proteomic and metabolomic analyses, and autoantibody pro-files.
An integrative analysis of these data highlights dynamic changes and
provides information about healthy and diseased phenotypes.
Whole-Genome Sequencing
We first generated a high quality genome sequence of this individual
using a variety of different technologies. Genomic DNA was subjected to
deep WGS using technologies from Complete Genomics (CG, 35 nt
paired end) and Illumina (100 nt paired end) at 150-and 120-fold total cov-erage,
respectively, exome sequencing using three different technologies
to 80-to 100-fold average coverage (see Extended Experimental Proce-dures
available online) and analysis using genotyping arrays and RNA se-quencing.
The vast majority of genomic sequences (91%) mapped to the hg19
(GRCh37) reference genome. However, because of the depth of our se-quencing,
we were able to identify sequences not present in the reference
sequence. Assembly of the unmapped Illumina sequencing reads
(60,434,531,9% of the total) resulted in 1,425 (of 29,751) contigs (span-ning
26 Mb) overlapping with RefSeq gene sequences that were not an-notated
in the hg19 reference genome. The remaining sequences
appeared unique, including 2,919 exons expressed in the RNA-Seq data
(e.g., Figure S1A). These results confirm that a large number of undocu-mented
genetic regions exist in individual human genome sequences and
can be identified by very deep sequencing and de novo assembly [34].
Our analysis detected many single nucleotide variants (SNVs), small in-sertions
and deletions (indels) and structural variants (SVs; large inser-tions,
deletions, and inversions relative to hg19), (summarized in Table 1
and Experimental Procedures). 134,341 (4.1%) high-confidence SNVs
are not present in dbSNP, indicating that they are very rare or private to
the subject. Only 302 high-confidence indels reside within RefSeq protein
coding exons and exhibit enrichments in multiples of three nucleotides
(p<0.0001). In addition to indels, 2,566 high-confidence SVs were identi-fied
(Experimental Procedures and Table S1) and 8,646 mobile element
insertions were identified [47].
Analysis of the subject’s mother’s genome by comprehensive genome se-quencing
(as above) and imputation allowed a maternal/paternal chromo-somal
phasing of 92.5% of the subject’s SNVs and indels (see Extended
Experimental Procedures for details). Of 1,162 compound heterozygous
mutations in genes, 139 contain predicted compound heterozygous dele-terious
and/or nonsense mutations. Phasing enabled the assembly of a
personal genome sequence of very high confidence (c.f., [41]).
WGS-Based Disease Risk Evaluation
We identified variants likely to be associated with increased susceptibility
to disease [12]. The list of high confidence SNVs and indels was analyzed
for rare alleles (<5% of the major allele frequency in Europeans) and for
changes in genes with known Mendelian disease phenotypes (data sum-marized
in Table 2), revealing that 51 and 4 of the rare coding SNV and
indels, respectively, in genes present in OMIM are predicted to lead to
loss-of-function (Table S2A). This list of genes was further examined for
medical relevance (Table S2A; example alleles are summarized in Figure
2A), and 11 were validated by Sanger sequencing. High interest genes in-clude:
(1) a mutation (E366K) in the SERPINA1 gene previously known in
the subject, (2) a damaging mutation in TERT, associated with acquired
aplastic anemia [57], and (3) variants associated with hypertriglyceridemia
and diabetes, such as GCKR (homozygous) [54], and KCNJ11 (homozy-gous)
[18] and TCF7 (heterozygous) [13].
Genetic disease risks were also assessed by the RiskOGram algorithm,
which integrates information from multiple alleles associated with disease
risk [6] (Figure 2B). This analysis revealed a modest elevated risk for
coronary artery disease and significantly elevated risk levels of basal cell
Table 1. Summary and Breakdown of DNA Variants
Type Total Variants Total High
Confidence
Heterozygous
High Confidence
Homozygous
High Confidence
Total SNVs 3,739,701 3,301,521 1,971,629 1,329,892
Total gene-associated SNVs 1,312,780 1,183,847 717.485 466.362
Total coding/UTR 49.017 44.542 27.383 17.159
Missense 10.592 9.683 5.944 3.739
Nonsense 83 73 49 24
Synonymous 11.459 10.864 6.747 4.117
5´UTR 4.085 2.978 1.802 1.176
3´UTR 22.798 20.944 12.841 8.103
Intron 1,263,763 1,139,305 690.102 449.203
Ts/Tv — 2.14 — —
dbSNP 3,493,748 3,167,180 — —
Candidate private SNV 245.953 134.341 — —
Indels (-107~ +36 bp) 1,022,901 216.776 — —
Coding 3.263 302 — —
Structural variants (>50 bp) 44.781 2.566 — —
In 1000G projecta 4.434 1.967 — —
High confidence values are from variants identified across multiple plat-forms
(Illumina and CG) and/or Exome and RNA-Seq data. Annotations
were based from variant call formatted (vcf) files for heterozygous calls:
0/1, reference (ref)/alternative (alt); 1/2, alt/alt and homozygous calls; 1/1,
alt/alt; 1/, (alt/alt-incomplete call). Polyphen-2 was used to identify the lo-cation
of the SNVs. a1000G (1000 Genomes Project Consortium, 2010).](https://image.slidesharecdn.com/skbiomed03-10-141201073043-conversion-gate01/85/Sk-biomed-03-10-54-320.jpg)
![МИРОВОЙ ОПЫТ
Figure 1. Summary of Study
(A) Time course summary. The subject was monitored for a total of 726 days,
during which there were two infections (red bar, HRV; green bar, RSV). The
black bar indicates the period when the subject: (1) increased exercise, (2) in-gested
54
81 mg of acetylsalicylic acid and ibuprofen tablets each day (the latter
only during the first 6 weeks of this period), and (3) substantially reduced
sugar intake. Blue numbers indicate fasted time points.
(B) iPOP experimental design indicating the tissues and analyses involved in
this study.
(C) Circos [26] plot summarizing iPOP. From outer to inner rings: chromosome
ideogram; genomic data (pale blue ring), structural variants >50 bp (deletions
[blue tiles], duplications [red tiles]), indels (green triangles); transcriptomic data
(yellow ring), expression ratio of HRV infection to healthy states; proteomic data
(light purple ring), ratio of protein levels during HRV infection to healthy states;
transcriptomic data (yellow ring), differential heteroallelic expression ratio of al-ternative
allele to reference allele for missense and synonymous variants (pur-ple
dots) and candidate RNA missense and synonymous edits (red triangles,
purple dots, orange triangles and green dots, respectively).](https://image.slidesharecdn.com/skbiomed03-10-141201073043-conversion-gate01/85/Sk-biomed-03-10-55-320.jpg)
![Партнер Фонда «Сколково»
55
Table 2. Summary of Disease-Related Rare Variants
Category Count
Total high confidence rare SNVs 289,989
Coding 2,546
Missense 1,320
Synonymous 1,214
Nonsense 11
Nonstop 1
Damaging or possibly damaging 233
Putative loss-of-function SNVsa* 51
Total high confidence rare indels 51,248
Coding indels 61
Frameshift indels 27
miRNA indels 3
miRNA target sequence indels 5
Putative loss-of-function indels* 4
carcinoma (Figure 2B), hypertriglyceridemia, and type 2 diabetes (T2D)
(Figures 2B and 2C).
In addition to coding region variants we also analyzed genomic variants
that may affect regulatory elements (transcription factors [TF]), which had
not been attempted previously (Data S1). A total of 14,922 (of 234,980)
SNVs lie in the motifs of 36 TFs known to be associated with the binding
data (see Experimental Procedures), indicating that these are likely hav-ing
a direct effect on TF binding. Comparison of SNPs that alter binding
patterns of NFkB and Pol II sites [24], also revealed a number of other in-teresting
regulatory variants, some of which are associated with human
disease (e.g., EDIL) [48] (Figure S1B).
Medical Phenotypes Monitoring
Based on the above analysis of medically relevant variants and the
RiskOGram, we monitored markers associated with high-risk disease
phenotypes and performed additional medically relevant assays.
Monitoring of glucose levels and HbA1c revealed the onset of T2D as di-agnosed
by the subject’s physician (day 369, Figures 2A and 2C). The
subject lacked many known factors associated with diabetes (nonsmoker;
BMI = 23.9 and 21.7 on day 0 and day 511, respectively) and glucose lev-els
were normal for the first part of the study. However, glucose levels ele-vated
shortly after the RSV infection (after day 301) extending for several
months (Figure 2D). High levels of glucose were further confirmed using
glycated HbA1c measurements at two time points (days 329, 369) during
this period (6.4% and 6.7%, respectively). After a dramatic change in diet,
exercise and ingestion of low doses of acetylsalicylic acid a gradual de-crease
in glucose (to ~93 mg/dl at day 602) and HbA1c levels to 4.7%
was observed. Insulin resistance was not evident at day 322. The patient
was negative for anti-GAD and anti-islet antibodies, and insulin levels cor-related
well with the fasted and nonfasted states (Figure S2C), consistent
with T2D. These results indicate that a genome sequence can be used to
estimate disease risk in a healthy individual, and by monitoring traits as-sociated
with that disease, disease markers can be detected and the phe-notype
treated.
The subject contained a TERT mutation previously associated with aplas-tic
anemia [57]. However, measurements of telomere length suggested lit-tle
or no decrease in telomere length and modest increase in numbers of
cells with short telomeres relative to age-matched controls (Figures S2A
and S2B). Importantly, the patient and his 83-year-old mother share the
same mutation but neither exhibit symptoms of aplastic anemia, indicating
that this mutation does not always result in disease and is likely context
specific in its effects.
Consistent with the elevated hypertriglyceridemia risk, triglycerides were
found to be high (321 mg/dl) at the beginning of the study. These levels
were reduced (81–116 mg/dl) after regularly taking simvastatin (20
mg/day).
We also examined the variants for their potential effects on drug response
(see Extended Experimental Procedures). Among the alleles of interest,
(Figure 2A and Table S2B) two genotypes affecting the LPIN1 and
SLC22A1 genes were associated with favorable (glucose lowering) re-sponses
to two diabetic drugs, rosiglitazone and metformin, respectively.
We followed the levels of 51 cytokines along with the C-reactive protein
(CRP) using ELISA assays, which revealed strong induction of proinflam-matory
cytokines and CRP during each infection (Figures 2E and 2F). We
also observed a spike of many cytokines at day 12 after the RSV infection
(day 301 overall). These data define the physiological states and serve as
a valuable reference for the omic profiles integrated into a longitudinal
map of healthy and diseased states described in the next sections.
Dynamic Omics Analysis: Integrative Omics Profiling
of Molecular Responses
We profiled the levels of transcripts, proteins, and metabolites across the
HRV and RSV infections and healthy states using a variety of ap-proaches.
RNA-Seq of 20 time points generated over 2.67 billion uniquely
mapped 101b paired-end reads (123 million reads average per time
point) and allowed for an analysis of the molecular complexity of the tran-scriptome
in normal cells (PBMCs) at an unprecedented level. The rela-tive
levels of 6,280 proteins were also measured at 14 time points through
differential labeling of samples using isobaric tandem mass tags (TMT),
followed by liquid chromatography and mass spectrometry (LC-MS/MS)
[10, 49]. A total of 3,731 PBMC proteins could be consistently monitored
across most of the 14 time points (see Figure S3A and Data S3). In addi-tion,
6,862 and 4,228 metabolite peaks were identified for the HRV and
RSV infection, and a total of 1,020 metabolites were tracked for both in-fections
(see Figure S4 and Data S4, [3]). Finally, as described below, we
also analyzed miRNAs during the HRV infection.
This wealth of omics information allowed us to examine detailed dynamic
trends related directly to the physiological states of the individual and re-vealed
enormous changes in biological processes that occurred during
healthy and diseased states. For each profile (transcriptome, proteome,
metabolome), we systematically searched for two types of nonrandom
patterns: (1) correlated patterns over time and (2) single unusual events
(i.e., spikes that may occur at any given time point defined as statistically
significantly high or low signal instances compared to what would be ex-pected
by chance). To perform this analysis, we developed a general
scheme for integrated analysis of data (see Figure S5 and Extended Ex-perimental
Procedures for further details). We used a Fourier spectral
analysis approach that both normalizes the various omics data on equal
basis for identifying the common trends and features, and, also accounts
for data set variability, uneven sampling, and data gaps, in order to detect
real-time changes in any kind of omics activity at the differential time
points (see Supplemental Information). Autocorrelations were calculated
to assess nonrandomness of the time-series (p<0.05 one-tailed based on
simulated bootstrap nonparametric distribution by sampling with replace-ment
of the original data, n>100,000), with significant signals classified as
autocorrelated (I). The remaining data was searched for spike events,
which were classified as spike maxima (II) or spike minima (III) (p<0.05
one-tailed based on differences from simulated, n>100,000 random distri-bution
of the time-series). After classification, the data were agglomerated
into hierarchical clusters (using correlation distance and average linkage)
of common patterns and biological relevance was assessed through GO
[5] analysis (Cytoscape [44], BiNGO [36] p<0.05, Benjamini-Hochberg [7]
adjusted p<0.05) and pathway analysis (Reactome [11] functional interac-tion
[FI], networks including KEGG [23, 44], p<0.05, FDR<0.05). The uni-fied
framework approach was implemented on all the different data sets
*In curated Mendelian disease genes.](https://image.slidesharecdn.com/skbiomed03-10-141201073043-conversion-gate01/85/Sk-biomed-03-10-56-320.jpg)
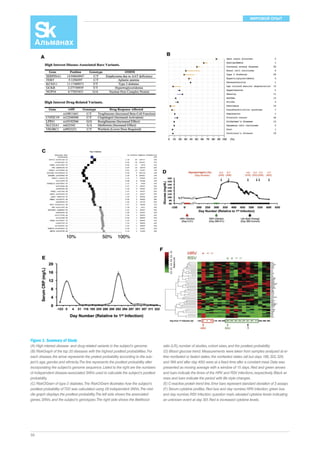
![Партнер Фонда «Сколково»
57
both individually and in combination, and our results revealed a number of
differential changes that occurred both during infectious states and the
varying glucose states.
We first analyzed the different individual transcriptome, proteome (serum
and PBMC) and metabolome data sets; the proteome and metabolome
results are presented in the Supplemental Information (Figures S3, S4,
S6 and Data S3-S6). A total of 19,714 distinct transcript isoforms [55] cor-responding
to 12,659 genes (Figure S1C) were tracked for the entire time
course, and their dynamic expression response was classified into either
autocorrelated (I) and spike sets, further subdivided as displaying maxima
(II) or minima (III) (Figure 3). The clustering and enrichment analysis dis-played
a number of interesting pathways in each class. In the autocorre-lated
group (Figure 3B, [I]; see also Figure S6A and Data S6, [1 and 2]),
we found two main trends: an upward trend (2,023 genes), following the
onset of the RSV infection, and a similar coincidental downward trend
(2,207 genes). The upward autocorrelated trend revealed a number of
pathways as enriched (p<0.002, FDR<0.05), including protein metabo-lism
and influenza life cycle. Additionally, the downward autocorrelation
cluster showed a multitude of enriched pathways (p<0.008, FDR<0.05),
such as TCR signaling in naive CD4+ T cells, lysosome, B cell signaling,
androgen regulation, and of particular interest, insulin signaling/response
pathways. These different pathways, which are activated as a response
to an immune infection, often share common genes and additionally we
observe many genes hitherto unknown to be involved in these pathways
but displaying the same trend. Furthermore, we observed that the down-ward
trend, that began with the onset of the RSV infection and appeared
to accelerate after day 307, coincided with the beginning of the observed
elevated glucose levels in the subject.
In the dynamic spike class we again saw patterns that were concordant
with phenotypes (Figure 3B, [II] and [III]; see also Figure S6A and Data
S6, [3-14]). A set of expression spikes displaying maxima (547 genes),
that are common to the onset of both the RSV and HRV infections are as-sociated
with phagosome, immune processes and phagocytosis,
(p<1×10-4, FDR<6×10-3). Furthermore, a cluster that exhibits an elevated
spike at the onset of the RSV infection involves the major histocompatibil-ity
genes (p<7×10-4, Benjamini-Hochberg adjusted p<0.03). A large num-ber
of genes with a coexpression pattern common to both infections in the
time course have yet to be implicated in known pathways and provide
possible connections related to immune response. Finally, our spike class
displaying minima showed a distinct cluster (1,535 genes) singular to day
307 (day 14 of the RSV infection), associated with TCR signaling again,
TGF receptors, and T cell and insulin signaling pathways (p<0.02,
FDR<0.03). Overall, the transcriptome analysis captures the dynamic re-sponse
of the body responding to infection as also evidenced by our cy-tokine
measurements, and also can monitor health changes over long
periods of time, with various trends.
To further leverage the transcriptome and genome data, we performed an
integrated analysis of transcriptome, proteomic and metabolomics data
for each time point, observing how this corresponded to the varying physi-ological
states monitored as described in the above sections. Because of
the availability of many time points through the course of infection, we ex-amined
in detail the onset of the RSV infection, as well as extended our
complete dynamics omics profile during the times that our subject began
exhibiting high glucose levels. Figure 4 shows an integrated interpretation
of omics data (see also Figure S6B and Data S7), where all trends are
combined for each omics data set and the common patterns emerge pro-viding
complementary information. In addition to the common patterns ob-served
in our transcriptome analysis, new patterns emerged, some
unique to protein data, some to metabolite, and some common to all. In
particular we found the following interesting results: for autocorrelated
clusters we found the same trends as observed in the transcriptome, ad-ditionally
augmented with concordant protein expressions. Pathways
such as the phagosome, lysosome, protein processing in endoplasmic
reticulum, and insulin pathways emerged as significantly enriched
(p<0.002, FDR<0.0075), and showed a downward trend post-infection,
and further accelerated after 3 weeks following the initial onset of the RSV
infection (this cluster comprised of 1,452 transcriptomic and 69 proteomic
components, corresponding to 1,444 genes). The elevated spike class
showed a maxima cluster on day 18 post RSV infection (one time point
after the cytokine maximum), with enrichment in pathways such as the
spliceosome, glucose regulation of insulin secretion, and various path-ways
related to a stress response (p<1×10-4, FDR<0.02) — this cluster
included 1,956 transcriptomic, 571 proteomic and 23 metabolomic com-ponents,
corresponding to 2,344 genes. Even though current proteomic
information is more limited than the full transcriptome because it follows
fewer components, as evidenced in Figure 4 (II), several pathways, in-cluding
the glucose regulation of insulin secretion pathway, clearly
emerge from the proteomic information and would not have been ob-served
by only monitoring the transcriptome. Additionally, in this cluster
we find significant GO enrichment in splicing and metabolic processes
(p<6×10-47, Benjamini-Hochberg adjusted p<10-45). Furthermore, inspec-tion
of metabolites reveals 23 that show the same exact trend (i.e., spikes
at day 18 post RSV infection); at least one, lauric acid has been impli-cated
in fatty acid metabolism and insulin regulatory pathways [27]. Fi-nally,
we observe minima spikes as well, with yet another interesting
group on day 18, which showed downregulation in several pathways
(p<0.003, FDR<0.05), such as the formation of platelet plug. This cluster
displayed a high degree of synergy between the various omics data, com-prised
of 3,237 transcriptomic and 761 proteomic components correspon-ding
3,400 genes and 83 metabolomic components.
In summary, our integrated approach revealed a clear systemic response
to the RSV infection following its onset and postinfection response, includ-ing
a pronounced response evident at day 18 post RSV infection. A vari-ety
of infection/stress response related pathways were affected along with
those associated to the high glucose levels in the later time points, includ-ing
insulin response pathways.
Dynamic Omics Analysis: Extensive Heteroallelic Variation
and RNA Editing
The considerable amount of transcriptome and proteome data allowed us
to analyze and follow changes in allele-specific expression (ASE), splic-ing,
and editing at the RNA and protein levels during healthy and dis-eased
states.
Of the 49,017 genomic variants associated with coding or UTR regions
(Table 1), 12,785 (26%) were expressed in PBMCs (R40 read coverage;
Table S3). A total of 8,509 of the variants are heterozygous (1,113 mis-sense)
and the remainder (4,686; 684 missense) are homozygous. Eight
of the 83 nonsense mutations were expressed indicating that not all non-sense
mutations result in transcript loss.
The numerous heterozygous variants allowed an analysis of the
dynamics of differential ASE, (shrunk ratios, Experimental Procedures;
Figures 5A and S7B) in PBMCs during healthy and diseased states. We
found 497 and 1,047 genes that exhibited differential ASE during HRV
and RSV infection, respectively (posterior probability ≥0.75, beta-binomial
model; ≥40 reads, ≥7 time points); many of these are immune response
genes, e.g., PADI4 and PLOD1 (Figure 5B). Among the differential ASE
sites 100 and 218 were specific to HRV and RSV infected states, respec-tively
(Figures 5C and 5D). Differential ASE genes in the HRV compared
to healthy phase were enriched for those encoding SNARE vesicular
transport proteins (DAVID analysis; Benjamini p<0.05). Summing over all
computed ASE alternative to total ratios revealed that nonreference het-eroallelic
variants were expressed at 98% of reference variants. The ex-pression
of over 50 heterozygous variants, including some of the rare/
private SNVs (which form 0.72% of the genomic total), and differentially
expressed variants (SVIL and TRIM5), was confirmed by Sanger cDNA
sequencing and/or digital PCR [19] of cDNA (Figures 5B and S7). Overall,
these results demonstrate that differential ASE is pervasive in humans
and is particularly distinct during healthy and infected states, with many of
these changes residing in immune response genes.
The depth of our RNA-Seq data enabled us to re-evaluate the extent of](https://image.slidesharecdn.com/skbiomed03-10-141201073043-conversion-gate01/85/Sk-biomed-03-10-58-320.jpg)
![МИРОВОЙ ОПЫТ
Figure 3. Transcriptome Time Course Analysis
(A) Summary of approach for identification of differentially expressed components.
The various omics sets were processed through a common framework involving
spectral analysis, clustering, and pathway enrichment analysis.
(B) Pattern classification. The different emergent patterns from the analysis of the
transcriptome for the entire time course are displayed for the autocorrelation (I),
58
spike maxima (II), and spike minima (III) classes. For different clusters, examples of
gene connections in selected pathways based on Reactome [11] FI (Cytoscape
plugin [44]) are shown as networks. Example GO [5] enrichment analysis results
from Cytoscape [44] BiNGO [36] plugin and pathway enrichment results (Reac-tome
FI [11]) are included.](https://image.slidesharecdn.com/skbiomed03-10-141201073043-conversion-gate01/85/Sk-biomed-03-10-59-320.jpg)
![Партнер Фонда «Сколково»
59
Figure 4. Integrated Omics Analysis
For days 186-400, the different emergent patterns from an integrated analysis of
the transcriptome, proteome, and metabolome data are displayed for the autocor-relation
(I), spike maxima (II), and spike minima (III) classes. For different clusters,
examples of gene connections in selected pathways based on Reactome [11]
and FI Cytoscape [44] plugin are shown as networks, with constituents marked as
assessed from proteome data, transcriptome data or both. Example GO [5] en-richment
analysis results from Cytoscape [44] BiNGO [36] plugin and pathway
enrichment results (Reactome FI [11]) are included.](https://image.slidesharecdn.com/skbiomed03-10-141201073043-conversion-gate01/85/Sk-biomed-03-10-60-320.jpg)
![МИРОВОЙ ОПЫТ
RNA editing (Figure 6 and Data S8 and S11A), typically an adenosine to
inosine (A-to-I) conversion [32] or infrequently cytidine to uridine (C-to-U),
in normal human cells. We found 2,376 high-confidence coding-associ-ated
60
RNA edits, including 795 A-to-I (A-to-G) and 277 C-to-U deamina-tion-
like edits (Figure 6A). A total of 587 edits in 175 genes were predicted
to cause amino acid substitutions (Polyphen-2 [3]); the remainder were
nonsense (11), synonymous (435), or located in 50/30 UTRs (103/1,240).
Ten edited bases causing amino acid substitutions were validated by
Sanger cDNA sequencing and/or digital droplet PCR, as well as by identi-fication
of their peptide counterparts by mass spectrometry (Figure 6B).
Interestingly, we identified A-to-G edits (Figure 6B), e.g., IGFBP7, BLCAP,
and AZIN1 in PBMCs that were known to occur in other tissues [16, 30],
indicating that the same RNA can be edited in other cell types. BLCAP
exhibited two edited changes (Figure 6C) with edited/total ratios of 0.12-
0.2 and 0.18-0.31, respectively, comparable to the 0.21 ratio previously
observed in the brain [14].
Furthermore, we found and validated two missense-causing edits, U-to-C
in SCFD2 and G-to-A in FBXO25 (Figure 6D), indicating an amination-like
RNA-editing mechanism, previously not observed in human cells. Our re-sults
reveal that a large number of edits occur and exhibit dynamic and
differential changes in populations of PBMCs (Figure 6B). The total num-ber
of edited RNAs, while extensive, is significantly lower than that re-ported
in human lymphoblastoid lines and very different in its distribution
[33]. We believe that in addition to tissue-specific variation, the observed
differences are also likely due to overcalling of false-positive SNVs, a
problem we corrected with deep exome sequencing, removal of repeat
regions and pseudo-genes, and strings of close-proximity variants (Data
S11A).
Finally, to determine whether the nonreference allele and edited RNAs
serve as templates for protein synthesis, we generated proteome data-bases
for 4,586 missense SNVs and all 30,385 edits and used them to
search our mass spectra from the untargeted protein profiling experi-ments
as well as in a targeted approach to directly search for 500 edited
proteins (see Extended Experimental Procedures). Peptides for 48 SNVs
Figure 5. Heteroallelic Expression
Study of PBMCs
(A) Frequency of allele-specific expression
(ASE) based on shrunk alternative/total ra-tios
of RNA-Seq data. A total of 143 posi-tions
fall outside the three standard
deviations (s) range (see Figure S7B;
<0.33, >0.66), suggesting that certain het-erozygous
alleles (DNA level) are preferen-tially
expressed in PBMCs. Standard
deviations (s) are denoted with dotted lines
and the average ratio overlapping across
all time points is 0.49.
(B) Digital droplet PCR validation of two
heteroallelic expressed genes PADI4 and
PLOD (relative to alternative allele).
(C) Heat map of the HRV infection time
course (seven time points) showing differ-ential
ASE during HRV infection day 0 (red
arrow) relative to average shrunk ratios of
healthy states (days 116-255).
(D) Heat map of the RSV infection time
course (13 time points) showing differential
ASE specific to RSV infection day 289 (red
arrow) relative to average shrunk ratios of
healthy states (days 311-400), onset of high
glucose on day 307 is also shown (red
arrow). Heat map ratios are relative to the
alternative allele (alternative/ total, posterior
probability >0.75). Example of enriched
KEGG pathway gene cluster [21] (Ben-jamini
p<0.05) shown below Figure 5C.
and 51 edits were identified (FDR<0.01 and requiring one unique peptide
per protein; Data S9 and S11B). A total of 17/17 selected SNVs (100%)
were validated by Sanger sequencing. Seven peptides derived from the
SNV and six peptides derived from edited transcripts were unique to a
single protein in the IPI database [25] and classified as high confidence.
These results indicate that a large fraction of personal variants are ex-pressed
as transcripts and a number of these are also translated as pro-teins.
miRNA Variant Analysis
In addition to the omics profiling above, we identified 619-681 known miR-NAs
from PBMCs per time point (>10 reads, days 4, 21, 116, 185, and
186), 106 of which showed dynamic changes (e.g., Figures S2D and
S2E). Examination of miRNA editing revealed 50 edited miRNAs (C-to-U
or A-to-I) with stringent criteria (edited reads >5% of total reads or >399
modified reads) indicating that at least 4% of expressed miRNAs are po-tentially
edited. Eighteen miRNAs contain edits located within the func-tionally
critical “seed sequences,” potentially affecting their mRNA targets.
Interestingly, expression of SNV-containing miRNAs was generally higher
compared to SNV-free miRNA (Figures 6E and 6F). In addition to edits,
analysis of the SNVs located in miRNAs revealed that most (25 of 31)
SNV-containing miRNAs were not expressed. These miRNAs were
among those discovered in cancer cell lines [22] and may not normally be
highly expressed in PBMCs from healthy individuals.
DISCUSSION
To our knowledge, our study is the first to perform extensive personal
iPOP of an individual through healthy and diseased states. It revealed ex-tensive
complex and dynamic changes in the omics profiles, especially in
the transcriptomes, between healthy states and viral infections, and be-tween
nondiabetic and diabetic states. iPOP provides a multidimensional
view of medical states, including healthy states, response to viral infec-tion,
recovery, and T2D onset. Our study indicates that disease risk can](https://image.slidesharecdn.com/skbiomed03-10-141201073043-conversion-gate01/85/Sk-biomed-03-10-61-320.jpg)
![Партнер Фонда «Сколково»
61
be assessed from a genome sequence and illustrates how traits associ-ated
with disease can be monitored to identify varying physiological
stages. We show that large numbers of molecular components are pres-ent
in blood samples and can be measured (>3 billion measurements
taken over 20 time points). For the transcriptome many of these arise
from differential splicing, ASE, and editing events. By observing dynamic
molecular changes that correspond to physiological states, this proof-of-principle
study offers a pilot implementation of personalized medicine. The
information obtained may greatly help in the design and application of
personalized health monitoring, diagnosis, prognosis, and treatment.
We speculate that differential expression of ASE/edits may be important
in monitoring and assessing diseased states. In this respect the
genes/proteins in which one isoform is abundant in one condition (e.g.,
diseased or healthy state) whereas another is abundant in another (e.g.,
diseased state) may provide unique physiological advantages to the indi-vidual
in distinct environmental conditions. Because multiple genes in our
study that exhibit ASE and editing changes are involved in immune func-tion,
we speculate that these components are particularly valuable for me-diating
immune responses to environmental conditions such as exposure
to pathogens. Likewise miRNA SNVs and edits, which also undergo dif-ferential
expression, may confer unique biological responses.
Although we analyzed a single individual, insights were gained by inte-grating
the multiple omics profiles associated with distinct physiological
states. Through examination of molecular patterns, clear signatures of dy-namic
biological processes were evident, including immune responses
during infection, insulin signaling response alterations after the RSV infec-tion.
Indeed, careful monitoring of omics changes across multiple time
points for the same individual revealed detailed responses, which might
not have been evident had the analyses been performed on groups due
to interindividual variability. Hence, we expect that our longitudinal person-alized
profiling approach provides valuable information on an individual
basis.
We focused on a generally healthy subject who exhibited no apparent dis-ease
symptoms. This is a critical aspect of personalized medicine, which
is to perform iPOP and evaluate the importance and changes of all the
profiles in ordinary individuals. These results have important implications
and suggest new paradigm shifts: first, genome sequencing can be used
to direct the monitoring of specific diseases (in this study, aplastic anemia
and diabetes) and second, by following large numbers of molecules a
more comprehensive view of disease states can be analyzed to follow
physiological states.
Our study revealed that many distinct molecular events and pathways are
activated both through viral infection and the onset of diabetes. Indeed,
the monitoring of large numbers of different components revealed a
steady decrease of insulin-related responses that are associated with dia-betes-
insulin response pathways occurring from the early healthy state to
a high glucose state. Although many of the activated and repressed path-ways
could be detected through transcript profiling, some were detected
only with the proteomics data and some with the combined set of data. In
addition a large number of connections with diabetes and insulin signaling
using metabolites, miRNAs, and autoantibodies were observed. One par-ticularly
interesting response detected with the proteomics data was the
onset of the elevated glucose response that was tightly associated with
the RSV infection and a particular subclinical response at day 12/18
postinfection. It is tempting to speculate that the RSV infection and/or the
associated event at day 12/18 triggered the onset of high glucose/T2D.
Although viral infections have been associated with T1D [52]), we are un-aware
of viral infection associated with T2D. Inflammation and activated
innate immunity have been associated with T2D (Pickup, 2004), and we
speculate that perhaps RSV triggered aberrant glucose metabolism
through activation of a viral inflammation response in conjunction with a
predisposition toward T2D. Although this cannot be proven with the analy-ses
from a single individual, this study nonetheless serves as proof-of-principle
that iPOP can be performed and provide valuable information.
Because diabetes is a complex disease there may be many ways to ac-quire
high glucose phenotype; longitudinal iPOP analysis of a large num-ber
of individuals may be extremely valuable to dissecting the disease
and its various subtypes, as well providing information into the molecular
mechanism of its onset.
Finally, we believe that the wealth of data generated from this study will
serve as a valuable resource to the community in the developing field of
personalized medicine. A large database with the complete time-dynamic
profiles for more individuals that acquire infections and other types of dis-eases
will be extremely valuable in the early diagnostics, monitoring and
treatment of diseased states.
EXPERIMENTAL PROCEDURES
The subject and mother in this study were recruited under the IRB proto-col
IRB-8629 at Stanford University. Full methods and associated refer-ences
can be found in the Extended Experimental Procedures section.
WGS was performed at Complete Genomics and Illumina. High-confi-dence
SNVs were mostly correct as evidenced by: (1) Illumina Omni1-
Quad genotyping arrays (99.3% sensitivity), (2) a Ti/Tv ratio of 2.14 as
expected (1000 Genomes Project Consortium, 2010), (3) Illumina capture
and DNA sequencing (92.7% accuracy), and (4) Sanger sequencing of 36
randomly selected SNVs (36/36 validated, Table S1). In contrast, the low
confidence SNVs had a Ti/Tv of only 1.46 and an accuracy of 63.8% (19
of 33 confirmed by Sanger sequencing, Table S1A). Similarly, the majority
of the 216,776 high-confidence indels are likely to be correct as (1)
Sanger sequencing validated 14 of 15 (93%) tested indels and (2)
exome-sequencing validated most indels (4,706, 82%); meanwhile the
806,125 low confidence indels had a low validation rate (5,225, 0.65%).
SVs were called using: (1) paired-end mapping [9] (2) read depth [2], (3)
split reads [59], and (4) junction mapping [28] to the breakpoint junction
database from the 1000 G [37]. A total of 2,566 were found by two differ-ent
methods or platforms (CG or Illumina) and were called high confi-dence;
>90% of these were in the database of genome variants.
Strand-specific RNA-Seq libraries were prepared as described previously
[39] and sequenced on 1-3 lanes of Illumina’s HiSeq 2000 instrument.
The TopHat package [50] was used to align the reads to the hg19 refer-ence
genome, followed by Cufflinks for transcript assembly and RNA ex-pression
analysis [50]. The Samtools package [31] was used to identify
variants including single nucleotide variants (SNV) and Indels. Small
RNAs were prepared from PBMCs for the first five time points; sequenc-ing
was performed according to Illumina’s Small RNA v1.5 Sample Prepa-ration
Guide.
The Luminex 51-plex Human Cytokines assay was performed at the
Stanford Human Immune Monitoring Center. For mass spectrometry, pro-teins
were prepared from PBMC cell lysates, labeled at lysines using the
TMT isobaric tags by Pierce, and digested with trypsin and analyzed
using reverse phase LC coupled to a Thermo Scientific (LTQ)-Orbitrap
Velos instrument. In order to profile serum, 14 major glycoproteins were
first removed using the Agilent Human 14 Multiple Affinity Removal Sys-tem
(MARS) column in order to analyze the less abundant constituents.
Metabolites were extracted by four times serum volume of equal mixture
of methanol, acetonitrile, and acetone and separated using our Agilent
1260 liquid chromatography. Hydrophobic molecules were profiled using
reversed phase UPLC followed by APCI-MS and hydrophilic molecule
were analyzed using HILIC UPLC followed by ESI-MS in either the posi-tive
or negative mode.
For the integrated analysis, per omics set, for each time-series curve the
Lomb-Scargle transformation [20, 35, 42, 43] for unevenly sampled
gapped time-series data was implemented [4, 15, 53, 58, 60]. This al-lowed
us to obtain a periodogram, which was used to calculate autocorre-lations
and then reconstruct the time-series with even sampling, allowing
standard time-series analysis and performing data clustering, while taking
the time intervals into account (see Extended Experimental Procedures).
Autoantibodyome profiling was performed using the Invitrogen ProtoArray
Protein Microarray v5.0 according to the manufacturer’s instructions.](https://image.slidesharecdn.com/skbiomed03-10-141201073043-conversion-gate01/85/Sk-biomed-03-10-62-320.jpg)




Документ представляет собой альманах, посвященный омиксным технологиям и их применению в России, исследуя их влияние на биомедицинскую науку и практику. Описываются достижения геномики и других омиксных направлений, а также перспективы их развития в подошедшие к новому уровню медицинских исследований и производстве лекарств. Авторы представляют мнения экспертов из разных областей, подчеркивая высокое качество работы российских специалистов в сфере биоинформатики и омиксных технологий.





















































