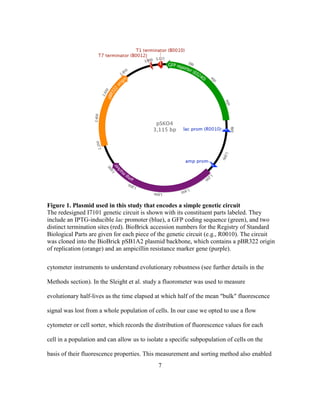
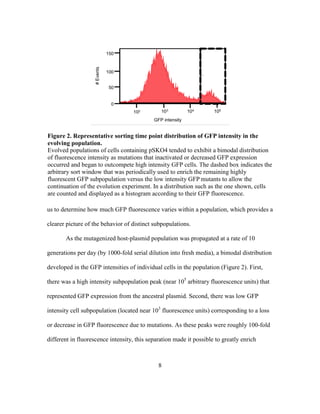
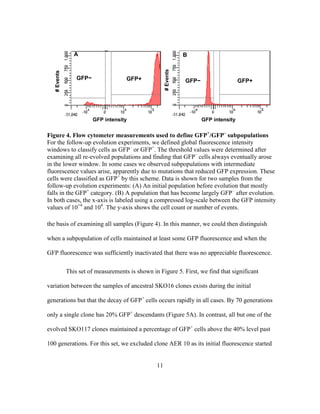
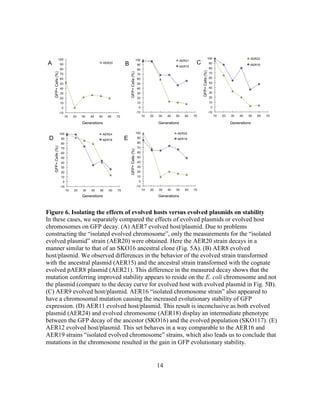
This document describes a thesis submitted by Alvaro Eugenio Rodríguez Mendoza to the University of Texas at Austin in partial fulfillment of the requirements for a Master of Arts degree. The thesis aimed to identify mutations that could enhance the evolutionary stability of green fluorescent protein expression from a plasmid in Escherichia coli. An evolution experiment was performed where a mutagenized E. coli population expressing GFP was periodically sorted by flow cytometry to select for cells with stable GFP expression over hundreds of generations. Genome sequencing of evolved strains with more stable GFP expression identified mutations in DNA polymerase genes PolA and PolB, which may enhance stability by lowering the mutation rate in the host.