Aims of thesession
Brief overview of the haematological system
Leukaemias
Lymphomas
Myelomas and other paraproteinaemias
SBA questions
4.
- Is agroup of malignant (neoplastic) disorders , characterized
by the clonal expansion and accumulation of one or more blood
cell line(s) , with eventual involvement of all hematopoietic
organs and other organs.
Leukemia
5.
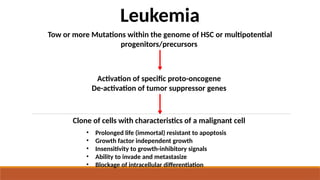
• Prolonged life(immortal) resistant to apoptosis
• Growth factor independent growth
• Insensitivity to growth-inhibitory signals
• Ability to invade and metastasize
• Blockage of intracellular differentiation
Tow or more Mutations within the genome of HSC or multipotential
progenitors/precursors
Activation of specific proto-oncogene
De-activation of tumor suppressor genes
Leukemia
Clone of cells with characteristics of a malignant cell
6.
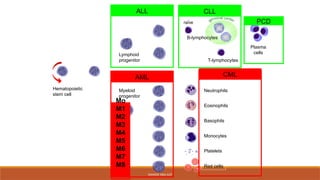
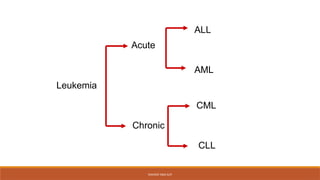
classification
◦ Leukemias areclassified into 2 major groups
◦ Chronic in which the onset is insidious, the
disease is usually less aggressive, and the cells
involved are usually more mature cells
◦ Acute in which the onset is usually rapid,
the disease is very aggressive, and the cells
involved are usually poorly differentiated with
many blasts.
9
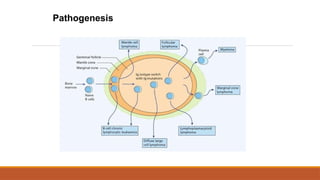
The genetics ofmalignant transformation
Malignant transformation - accumulation of genetic mutations in
cellular genes
The genes involved in the development of cancer are divided
broadly into two groups:
Oncogenes
Arise because of gain-of-function mutations in normal cellular
genes called proto-oncogenes
Oncogenic versions are generated when the activity of proto-
oncogenes is increased or they acquire a novel function through:
Translocation
Mutation
Duplication
Tumour suppressor genes
May acquire loss-of-function point mutation or deletion leading
to malignant transformation
10.
10
Leukemia cont’d
Although virusescause several forms of leukemia in animals,
their role in humans is uncertain
Only two viral associations are identified
Epstein-Barr virus, a DNA virus, associated with Burkitt's lymphoma
Human T-cell lymphotropic virus type I, called human T-cell leukemia/lymphoma virus, an RNA retrovirus,
associated with some T-cell leukemias and lymphomas
Exposure to ionizing radiation and certain chemicals (e.g.,
benzene, some anti-neoplastic drugs) is associated with an
increased risk of leukemia
11.
11
Leukemia cont’d
Some geneticdefects (e.g., Down syndrome, Fanconi's anemia)
also predispose to leukemia
Classification of leukemia
The main classification is into acute and chronic leukemia
On the basis of morphology and cytochemistry, acute leukemia
is further subdivided into:
Acute myeloid (myeloblastic/myelogenous) leukemia (AML)
Acute lymphoblastic (lymphocytic) leukemia (ALL)
AML is further subdivided into eight variants on a morphological
basis according to the French-American-British (FAB) scheme (M0
– M7)
12.
12
Classification of Leukemiacont’d
ALL is subdivided on a morphological basis according to the French-American-
British (FAB) classification into L1, L2, and L3
The chronic leukemias comprise two main types:
Chronic myeloid leukemia (CML)
Chronic lymphocytic (lymphatic) leukemia (CLL)
Other chronic types include:
Hairy cell leukemia
Prolymphocytic leukemia
Various leukemia/lymphoma syndromes
13.
Acute leukaemias
Patients presentwith pancytopaenia
◦ Symptoms of anaemia
◦ Infections
◦ Bleeding
◦ NB AML M3 presents with DIC!
Diagnosis to death 6-12 weeks if untreated
Chemosensitive
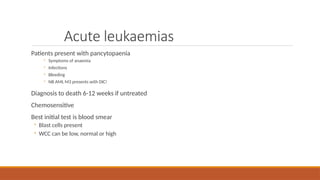
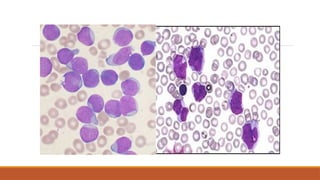
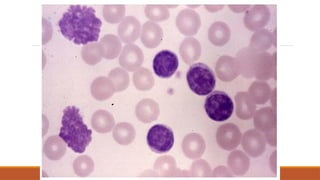
Best initial test is blood smear
◦ Blast cells present
◦ WCC can be low, normal or high
15.
Chronic leukaemias
Patients presentmore insidiously
◦ SBA tip – if patient presents at a ‘routine check-up’ answer is much more likely to be chronic leukaemia
◦ Less likely to present with pancytopaenia
Diagnosis to death 6-12 years if untreated
Less chemosensitive
Best initial test is full blood count
◦ WCC always high
◦ Look at white cell differential
‘Smudge’ cells in CLL on smear
17.
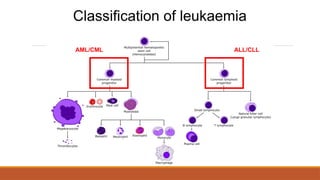
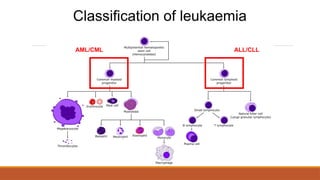
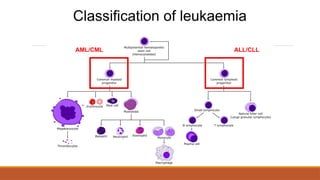
Classification of leukaemia
•Acutelymphoblastic leukaemia (ALL)
•Chronic lymphoblastic leukaemia (CLL)
•Acute myeloid leukaemia (AML)
•Chronic myeloid leukaemia (CML)
By cell type
20
The Acute Leukemias
Theleukemic cell population in ALL and AML probably result from clonal
proliferation by successive divisions form a single abnormal stem or progenitor
cell
There are over 50% myeloblasts or lymphoblasts in the bone marrow at clinical
presentation, and these blast cells fail to differentiate normally but are capable
of further divisions
Replacement of the normal hemopoietic precursor cells of the bone marrow
by myeloblasts or lymphoblasts and, ultimately in bone marrow failure
21.
21
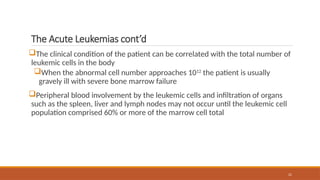
The Acute Leukemiascont’d
The clinical condition of the patient can be correlated with the total number of
leukemic cells in the body
When the abnormal cell number approaches 1012
the patient is usually
gravely ill with severe bone marrow failure
Peripheral blood involvement by the leukemic cells and infiltration of organs
such as the spleen, liver and lymph nodes may not occur until the leukemic cell
population comprised 60% or more of the marrow cell total
22.
22
The Acute Leukemiascont’d
The disease may be recognized by conventional morphology
only when blast (leukemic) cells in the marrow exceed 5% of the
cell total (unless the blast cells have some particular abnormal
feature, e.g., Auer rods in myeloblasts)
This corresponds to a total cell count in excess of 108
The clinical presentation and mortality in acute leukemia arises
mainly from:
Neutropenia
Thrombocytopenia, and
Anemia because of bone marrow failure
Organ infiltration, e.g., of he meninges or testes (less commonly)
•Imaging
-CXR, CT (pneumonia,mediastinal mass, lytic
bone lesions)
•BM biopsy
-Flow cytometry, cytogenetics and
immunohistochemistry
->30% blasts
26.
Acute myeloid leukaemia
Alsoknown as ‘myelogenous’.
Epidemiology
•Incidence = 3.7/100,000. Slight male
predominance.
•Commonest type of adult leukaemia (90%)
•Can occur at any age but median is 70 years
Prognosis of acuteleukaemia
5 year survival rates
•ALL
-Children- 75%
-Adults- 40%
-Worse if <1 or >60, high WBC, >4w to
remission
•AML
-30-50%
-Better if BM Tx, children, worse if >60
Prognosis of acute leukaemia
5 year survival rates
•ALL
-Children- 75%
-Adults- 40%
-Worse if <1 or >60, high WBC, >4w to
remission
•AML
-30-50%
-Better if BM Tx, children, worse if >60
Acute Lymphoid Leukemia
Malignantneoplastic proliferation and accumulation
of immature and nonfunctional Lymphoid line of
blood cells in the bone marrow
Acute lymphocytic leukemia is the most common
type of cancer in children
.
35.
Acute Lymphoid Leukemia
Signsand Symptoms
- Generalized weakness and fatigue
- Anemia
- Frequent or unexplained fever and infection
- Weight loss and/or loss of appetite
- Bone pain, joint pain (caused by the spread of "blast" cells
to the surface of the bone or into the joint from the marrow
cavity)
- Breathlessness
- Enlarged lymph nodes, liver and/or spleen
-Pitting edema (swelling) in the lower limbs and/or
abdomen
- Petechiae, which are tiny red spots or lines in the skin
36.
Acute Lymphoid Leukemia
-Damage to DNA that leads to uncontrolled cellular growth and
spread throughout the body.
Pathophysiology
- This damage may be caused by environmental factors such as
chemicals, drugs or radiation.
- Some evidence suggests that secondary leukemia can develop
in individuals treated for other cancers with radiation and
chemotherapy as a result of that treatment
- Damage can be caused through the formation of fusion genes,
as well as the dysregulation.
37.
Initial Laboratory findingcharacteristic of ALL
Peripheral Blood: 1- Leukocyte count usually increased but may be normal or decreased
.
2
-
Neutropenia
3
-
Lymphoblast
4
-
Thrombocytopenia
Bone Marrow: 1- Hypercellular
2
> -
20%
lymphoblast ( WHO)
Other laboratory finding: 1- associated with increased cellular metabolism and turn over such
:
Hyperuricemia
.
incrased serum LD
.
hypercalacemia due to increased BM resorption
.
2
-
Renal failure
.
3
-
increased CSF lymphblast
.
38.
Acute Lymphoid Leukemia
-Terminal Deoxynucleotidyl transferase (TdT):
- - TdT, is the most important enzyme that is helpful in the
identifying cellular subtypes.
- TdT is a DNA polymerase found in cell nuclei , this enzyme not
present in the normal mature lymphocyte but can be found in
65% of the thymic population of lymphocyte.
39.
Acute Lymphoid Leukemia
Classification
-Based on FAB classification, ALL is categorized into three
catergories ALL-L1, ALL-L2 and ALL-L3 .
- Immunohistochemistry and immunophenotyping are
almost always necessary to distinguish ALL from AML.
- On other hand WHO identify of malignant cell as T, B and
on the degree of maturation.
40.
Acute Lymphoid Leukemia
-Blasts in ALL-L1 are with high N/C ratio.
- Delicate diffuse chromatin pattern and small prominent
nucleoli.
- Scant cytoplasm .
- Affects primarily children
FAB classification
ALL-L1
42.
Acute Lymphoid Leukemia
FABclassification
ALL-L2
- The blasts are larger than those of L1, have more plentiful
cytoplasm and are more pleomorphic.
- Abundant cytoplasm, predominant nucleoli, nuclear
clefting .
-
Affects adults
44.
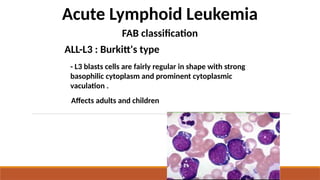
Acute Lymphoid Leukemia
FABclassification
ALL-L3 : Burkitt's type
- L3 blasts cells are fairly regular in shape with strong
basophilic cytoplasm and prominent cytoplasmic
vaculation .
Affects adults and children
45.
Findings specific forALL
•Examination
-Lymphadenopathy
-Splenomegaly (10-20% presentation)
-CNS signs- more likely
Bone and joint pain,easy bruising,fever,frequent
infections
•Bloods
-Anaemia- usually severe, signs present
-WBCs- variable, usually neutropenia
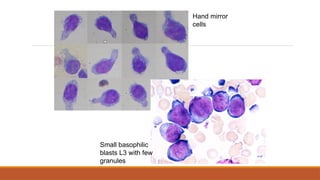
-Smear- smallish basophilic blasts, few
granules, hand-mirror cells
-Clotting- 10% ALL presents with DIC
•Imaging
-Mediastinal mass in some T cell ALL
Chronic lymphoblastic leukaemia
98%develop from B cells.
Epidemiology
•Incidence = 4.2/100,000. Slight male
predominance.
•Most common form of leukaemia in the West
•Usually >55, median age 72, rare <40.
Findings specific forCLL
Usually asymptomatic!
• Examination
-Lymphadenopathy/splenomegaly present in
late disease.
• Bloods
-WBCs- extremely high
-Smear- lymphocytosis with ‘smudge/basketball
cells’
• Other
-Richter’s syndrome
-Prolymphocytic transformation
55
Chronic lymphocytic leukemiacont’d
Laboratory findings in CLL
Lymphocytosis
The absolute lymphocyte count is >5 x 109
/l and may be up to
300x109
/l or more
Between 70% and 99% of white cells in the blood film appear
as small lymphocytes
Smudge or smear cells are also present
56.
56
Laboratory findings inCLL
Normocytic normocytic anemia is present in later states due to
marrow infiltration or hypersplenism
Autoimmune hemolysis may also occur
Thrombocytopenia occurs in many patients
Bone marrow aspiration shows lymphocytic replacement of normal
marrow elements
Lymphocytes comprise 25-95% of all the cells
Reduced concentrations of serum immunoglobulins
More marked with advanced disease
Rarely a paraprotien is present
57.
Chronic myeloid leukaemia
Stepped-upproduction of granulocytes and their
precursors and failed apoptosis leads to insidious
progression towards a blast crisis.
Epidemiology
•Incidence= 0.6-2/100,000
•Can occur at any age but rare in children. Peak
incidence at 40-60
•Less common than AML, CLL
58.
Pathophysiology
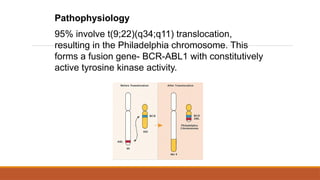
95% involve t(9;22)(q34;q11)translocation,
resulting in the Philadelphia chromosome. This
forms a fusion gene- BCR-ABL1 with constitutively
active tyrosine kinase activity.
59.
Findings specific forCML
Usually asymptomatic!
•Examination
-Splenomegaly- may be only feature at latent
phase, massive later on
•Bloods
-Anaemia- mild, worsens with progression
-WBCs- extremely high
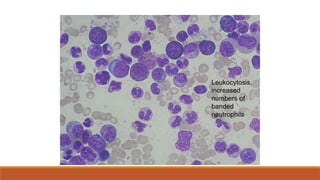
-Smear- leukocytosis with granulocyte left-shift
61
Chronic Myeloid Leukemia(CML) cont’d
It is an acquired abnormality of hemopoietic stem cells that is present in all dividing:
Granulocytic cells
Erythyroid cells
Megakaryocytic cells in the marrow
And also in some B and probably a minority of T lymphocytes
A great increase in total body granulocyte mass is responsible for most of the clinical
features
In at least 70% of patients there is a terminal metamorphosis to acute leukemia
(myeloblastic or lymphoblastic) with an increase of blast cells n the marrow to 50% or
more
62.
62
Chronic Myeloid Leukemia(CML) cont’d
It occurs in either sex (male: female = 1.4:1), most frequently
between the ages of 40 and 60 years
It may occur in children and neonates and in the very old
In most cases there are no predisposing factors but the
incidence was increased in survivors of the atom bomb exposures
in Japan
Laboratory findings in CML
Leucocytosis is usually >50x109
/l and sometimes >500x109
/l
A complete spectrum of myeloid cells is seen in the peripheral
blood
The levels of neutrophils and myelocytes exceed those of blast
cells and promyelocytes
63.
63
Laboratory findings inCML cont’d
Philadelphia (Ph) chromosome on cytogenetic analysis of blood
or bone marrow
Hypercellular bone marrow with granulopoietic predominance
Neutrophil alkaline phosphatase score is invariably low
Increased circulating basophils
Normochromic, normocytic anemia is usual
Platelet count may be increased (most frequently), normal or
decreased
Serum vitamin B12 and vitamin B12-binding capacity are
increased
Serum uric acid is usually raised
64.
Treatment of chronicleukaemia
CLL
•Watchful waiting with regular monitoring
•Chemotherapy. Indications:
-Severe systemic symptoms
-Non-pred-responsive AI
anaemia/thrombocytopenia
-Progressive splenomegaly/lymphadenopathy
-Increased WBC/reduced ‘doubling time’
Treatment of chronic leukaemia
65.
CML
•Imatinib
-Tyrosine kinase inhibitor,targets BCR-ABL1.
Greatly increases 5 year survival compared to
older drug therapies
-Initial treatment, continued indefinitely if optimal
response.
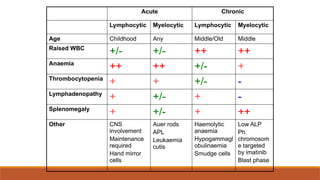
Acute Chronic
Lymphocytic MyelocyticLymphocytic Myelocytic
Age Childhood Any Middle/Old Middle
Raised WBC
+/- +/- ++ ++
Anaemia
++ ++ +/- +
Thrombocytopenia
+ + +/- -
Lymphadenopathy
+ +/- + -
Splenomegaly
+ +/- + ++
Other CNS
involvement
Maintenance
required
Hand mirror
cells
Auer rods
APL
Leukaemia
cutis
Haemolytic
anaemia
Hypogammagl
obulinaemia
Smudge cells
Low ALP
Ph
chromosom
e targeted
by imatinib
Blast phase
68.
Myeloproliferative neoplasms
Non leukaemicmalignancies
◦ Polycythaemia vera
◦ Essential thrombocythaemia
◦ Primary Myelofibrosis
◦ MaStocytosis-read fyi
Form part of a spectrum with CML
All are closely related to each other
Associated with JAK2 mutations
71.
Polycythaemia vera
Clinical features
◦Night sweats
◦ Hyperviscosity symptoms
◦ Pruritus after a hot bath
◦ Plethoric face
◦ Splenomegaly
◦ Haemorrhage
◦ Hypertension
◦ Gout
72.
Polycythaemia vera
Important labfeatures
◦ Increased Hb, HCT, Red cell mass
◦ Low serum EPO
◦ JAK2 mutation in 95%
Treatment
◦ Venesection
◦ Cytotoxic myelosuppression
◦ Hydroxyurea
◦ Busulphan
73.
Essential Thrombocythemia
A chronicmyeloproliferative neoplasm (MPN) characterized by persistent thrombocytosis
(platelet count >450 × 10⁹/L) due to clonal proliferation of megakaryocytes in the bone marrow.
Treatment hydroxyurea,aspirin
74.
Key Features
Feature Description
Bonemarrow
Hypercellular with increased
megakaryocytes (large, mature forms)
Common mutation
JAK2 V617F (50–60%), CALR (20–25%),
MPL (3–5%)
Blood findings Elevated platelet count, large platelets,
mild leukocytosis
Complications
Thrombosis (arterial or venous),
hemorrhage (due to dysfunctional
platelets), transformation to
myelofibrosis or AML
Treatment
Low-dose aspirin, hydroxyurea
(cytoreductive), interferon-α in young
or pregnant patients
75.
Primary Myelofibrosis
Primary Myelofibrosisis a chronic myeloproliferative neoplasm (MPN) characterized by:
Clonal proliferation of abnormal megakaryocytes and granulocytes in the bone marrow.
This leads to progressive bone marrow fibrosis, extramedullary hematopoiesis (blood cell
formation outside the marrow), and splenomegaly.
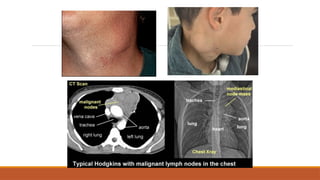
Hodgkin’s lymphoma
Originates fromB cells in the germinal centres of
lymphoid tissue and is characterised by orderly
spread from one LN group to another.
Epidemiology
•Incidence = 2.2/100,000, 30% of all lymphoma
•Bimodal distribution with peaks at 15-30 and >50
years
•Slight male predominance
Presentation
•Painless non-tender rubberyenlarged LN
-Cervical involvement in 60-70%, axillary in 10-
15%, inguinal in 6-12%
-May increase/decrease in size spontaneously
-May become ‘matted’ and non-mobile
-Contiguous progression to nearby groups
-Alcohol-induced pain
•Systemic symptoms
-Especially fever (30%), may be cyclic
-And severe pruritis (25%)
•Other
-Early satiety due to splenomegaly
Treatment
Definitive
•IA/IIA
-Radiotherapy alone- affectednodes and
prophylatically
-Chemo with radiotherapy of affected nodes
•IB/IIB/III/IV
-Chemo
•BM transplant
-If still progressive despite chemo or after
induction of remission after relapse
89.
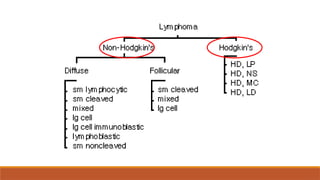
Non-Hodgkin’s lymphoma
A heterogeneousgroup of lymphoid tumours,
mostly of B cell origin,T cells, NK cells. No RS
cells.
Characterised by irregular pattern of spread and
common extranodal disease, they vary in their
aggressiveness.
Epidemiology
•Incidence = 17/100,000
•Median age is >50
•Diffuse large B cell and follicular commonest
90.
Risk factors
•Acquired
-Infection e.g.EBV (Burkitt’s, sinonasal), HTLV-1
(T cell), HCV, HHV8 (Kaposi’s), H. pylori (gastric
MALT)
-Previous chemotherapy/Hodgkin’s
-Autoimmune disorders e.g. Sjogren’s,
Hashimoto’s
-Immunodeficiency e.g. post-transplant, HIV/AIDS
Inherited
Presentation
•Painless non-tender rubberyenlarged LN
-Non-contiguous progression
•Systemic symptoms
-Commoner in high-grade
•Rash
-Cutaneous involvement e.g. mycosis fungoides, anaplastic large-
cell etc.
•Abdominal pain, early satiety
-Splenomegaly but unusual as rarely massive
-Hepatomegaly
•Mass
-Testicular
-GI, symptoms of obstruction
•Shortness of breath, pleuritic chest pain, SVC syndrome
-Mediastinal mass in high grade
•Neurological
-Primary CNS lymphoma, commoner in immunosuppressed
Treatment
•Low grade
-Localised (rare)-radiotherapy, surgery
-Disseminated- watch and wait or chemo when
symptomatic/organ dysfunction
-Gastric MALT- associated with H pylori,
antibiotic therapy curative in 90%
•High grade
-Aggressive chemo e.g. CHOP
-Maintenance not needed
-Allogenic stem cell transplantation
-CNS prophylaxis in very high grade e.g.
Burkitt’s
96.
Prognosis of lymphoma
5year survival rates
•Hodgkin’s- highly curable
-I/II- 90%
-IV- 65%
-Long-term sequelae of treatment
•Non-Hodgkin’s- vary widely (see IPI)
-Overall 63%
-Indolent follicular lymphoma I/II- 91% but may
not be curable
-DLBLC- curable with aggressive chemo
97.
Multiple Myeloma
Increased clonalplasma cells in the bone marrow
◦ >20% monoclonal plasma cells on bone marrow biopsy
Presdisposing factors
◦ Radiation
◦ Benzene
◦ Pesticides
Epidemiology
◦ 4 per 100,000 per year
◦ Median age 66 years
Pathophysiology
◦ Post germinal centre B cell proliferation
◦ Monoclonal antibody
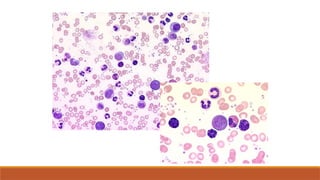
Ix: The myelomascreen
FBC and film
ESR
Urine dipstick
24 hour urine collection
U&Es
Urate
Albumin, calcium, phosphate, ALP
Serum and urinary electrophoresis
Serum Ig
X-ray
(BM Biopsy –diagnostic rather than screening)
108.
Myeloma: diagnosis
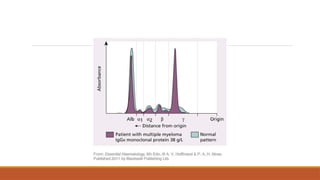
1. Productionof a single monoclonal antibody (paraprotein)
◦ ‘M’ band in γglobulin region on serum/ urine electrophoresis
2.Increased clonal plasma cells in the bone marrow
◦ >20% monoclonal plasma cells on bone marrow biopsy
3. Evidence of organ damage (‘CRAB HAI’)
110.
MM Management
Prognosis
◦ MMremains an incurable disease
◦ Mean survival 3-4 yrs from diagnosis
Treatment
◦ Specific
◦ Supportive
111.
Specific treatment
Intensive ornon intensive
◦ Intensive if <65
◦ Non intensive if >65
Intensive
◦ 4-6 cycles chemotherapy
◦ Cyclophosphamide, dexamethasone, thalidomide
◦ THEN autologous stem cell collection and transplant
Non-intensive
◦ Chemo: Melphalan and cyclophosphamide
MM- Summary
Post germinalcentre B cell proliferation
Monoclonal antibody/ paraprotein production
◦ M Band
◦ BJP
>20% monoclonal plasma cells in BM
‘CRAB HAI’
Specific and supportive treatment
Outcome still poor
A 66 yearold man has a FBC done whilst being
tested for hypercholesterolaemia and was found
to have a WBC of 15.4x109
/L with the rest of the
count otherwise normal. The most likely
diagnosis is:
a)Infectious mononucleosis
b)CMV infection
c)ALL
d)Pertussis
e)Chronic lymphocytic leukaemia
e)
116.
A 46 yearold woman presents with weight loss and
abdominal enlargement. She has also noticed
she is sweating more than normal and her
temperature is 38C. She is found to have
hepatosplenomegaly (liver 2cm below RCM,
spleen 6cm below LCM). Lymph nodes are not
enlarged. FBC shows: WBC 98x109
/L, Hb 8.3g/L,
Plts 504x109
/L.
A blood smear was performed.
118.
The blood smearshows increased numbers of
neutrophils, eosinophils and basophils. In
addition, there are increased numbers of
promyelocytes (but infrequent blast cells).
What is the most like disorder?
What is the optimum treatment for this patient?
a)Allogenic stem cell transplantation
b)Combination chemo
c)Imatinib
d)Blood transfusion to relieve symptoms
e)Rifampicin and isoniazid
c) (CML)
119.
Which ONE ofthese is the most likely clinical
presentation of a child with acute lymphoblastic
leukaemia?
a) A 6 month history of fatigue and repeated upper
respiratory tract infection
b) Poor appetite and abdominal pain resulting from
swollen spleen
c) Swollen gums in the mouth
d) Recent history of bruising and tiredness
e) None- incidental finding
120.
A 67 yearold lady is found to have an Hb of 18.9g/dL. Her erythropoeitin level is markedly
raised. Which of the following is the least likely diagnosis?
A) COPD
B) Eisenmenger’s syndrome
C) Polycythaemia vera
D) Renal cell carcinoma
E) Nepalese woman living at high altitude
121.
SBAs
A 67 yearold lady is found to have an Hb of 18.9g/dL. Her erythropoeitin level is markedly
raised. Which of the following is the least likely diagnosis?
A) COPD
B) Eisenmenger’s syndrome
C) Polycythaemia vera
D) Renal cell carcinoma
E) Nepalese woman living at high altitude
122.
Which ONE ofthese is NOT TRUE regarding the
Reed-Sternberg cell in Hodgkin’s lymphoma?
a) It is thought to be of B cell lineage
‐
b) It is multinucleate
c) It represents the majority of cells in a lymph
node of Hodgkin's lymphoma
d) It usually expresses CD15 and CD30
e) Their absence has a high negative predictive
value
c) Near-pathogonomic but in the minority of cells
123.
A 6 yearold boy from Kenya develops swelling of
the jaw. The mass responds rapidly to
chemotherapy. What is the most likely diagnosis?
a) Burkitt's lymphoma
b) Follicular lymphoma
c) Mycosis fungoides
d) Lymphoblastic lymphoma
e) Enteropathy-associated T cell lymphoma
a) Burkitt’s lymphoma occurs in the context of
chronic malaria infection causing reduced immunity
to EBV. Also associated with AIDS.
124.
SBAs
A 59 yearold man receiving chemotherapy for Non Hodgkin’s Lymphoma develops painful
haematuria. Which of the following is the most likely cause of his symptoms?
A) Rituximab
B) Cyclophosphamide
C) Adriamycin (doxorubicin/ hydroxydaunarubicin)
D) Vincristine (oncovin)
E) Prednisolone
125.
SBAs
A 59 yearold man receiving chemotherapy for Non Hodgkin’s Lymphoma develops painful
haematuria. Which of the following is the most likely cause of his symptoms?
A) Rituximab
B) Cyclophosphamide
C) Adriamycin (doxorubicin/ hydroxydaunarubicin)
D) Vincristine (oncovin)
E) Prednisolone
SBAs
A 63 yearold woman is about to commence chemotherapy for treatment of MM. Which of the
following medications should she be started on prior to chemotherapy?
A) Colchicine
B) Dexamethasone
C) Diclofenac
D) Allopurinol
E) Hydroxychloroquine
128.
SBAs
A 63 yearold woman is about to commence chemotherapy for treatment of MM. Which of the
following medications should she be started on prior to chemotherapy?
A) Colchicine
B) Dexamethasone
C) Diclofenac
D) Allopurinol
E) Hydroxychloroquine
129.
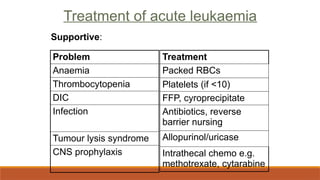
Tumour lysis syndrome
Malignantcells release intracellular contents after the first dose of chemotherapy
◦ Hyperkalaemia
◦ Hyperuricaemia
◦ Renal failure
◦ Allopurinol inhibits xanthine oxidase and prevents hyperuricaemia
◦ Large volumes of fluid should be given pre-chemo to prevent renal failure
130.
SBAs
A patient hasa sharp M band on serum electrophoresis. Which of the following is least
consistent with this result?
A) IgG MM
B) Waldenstrom’ macroglobulinaemia
C) AA amyloidosis
D) MGUS
E) CLL
131.
SBAs
A patient hasa sharp M band on serum electrophoresis. Which of the following is least
consistent with this result?
A) IgG MM
B) Waldenstrom’ macroglobulinaemia
C) AA amyloidosis
D) MGUS
E) CLL
#17 In reality it may not be possible to reliably distinguish lymphoblastic vs myeloid apart purely on presentation .However! We’ll teach you things which are more common in each to help you make an educated guess for SBAs.
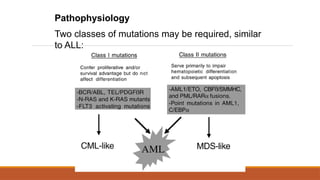
#23 Usual suspects e.g. radiation, benzene exposure (find a more comprehensive list on the handout) but importantly- being treated for leukaemia can make you more prone to getting leukaemia! Other haematological conditions that may involve ‘one hit’ or some degree of abnormal cell differentiation e.g. myelodysplasia can be viewed as ‘pre cancerous condition’ where impairment of differentiation leads to reduced production of RBC WBC Plts and develops into AML in 1/3 of cases.
Acquired
-Babies in nursery/day care have increased incidence of ALL
-Downs- x20 risk of ALL
#24 Things to look for in examination:
CV- make sure healthy! Some drugs cardiotoxic (anthracyclines). Flow murmur in anaemia
Resp- T-cell- mediastinal mass, infection
Abdo- Splenomegaly
Neuro- CNS involvement- headache, irritability, altered mental status, neck stiffness (cranial nerve III, IV, VI, VIII palsy in mature B-cell ALL)
Other- bruising, bleeding, temperature, lymphadenopathy, gum hypertrophy, skin infiltration
FBC- Failure of production of three types or, increased WBC (commoner in chronic). Neutropenia can occur regardless of high lymphocytes so a high white cell count doesn’t rule this out.
Clotting screen- 10% ALL present with DIC
U&Es- hyperuricaemia if large tumour burden-> renal failure
LDH usually raised due to increased cell turnover, also prognostic factor
#25 Mediastinal mass in some T cell ALL
Pneumonia due to neutropenia
BM biopsy
FAB- French-American-British. WHO use 20% instead.
Flow cytometry/cytogenetics- establish cell type (new WHO classification- B cell vs T cell, early B-precursor, pre-B cell, B cell) and translocation- targeted therapies, prognosis e.g. Philadelphia chromosome = bad, usually in adults, BCR-ABL may identify ALL arising from CML
#26 In adults, AML is commoner. Male predominance increases with age.
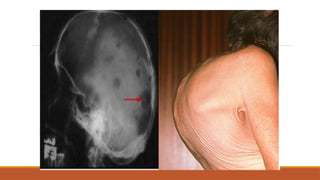
#28 Chloroma/myeloid- extra BM collection of myeloid leukaemia cells, overlap with leukaemia cutis, meningeal leukaemia, can be anywhere! Gum infiltration may occur
Hypokalaemia- lysozyme secretion affecting tubular activity
#31 Induction- aim to get into remission (<5% blasts in BM, normal blood cells, no blasts in blood, no symptoms/signs of disease)
Quadruple therapy- high-risk paeds and adult
BM Tx- e.g. Philadelphia chromosome in ALL, poor response to initial treatment, relapse in high risk ALL. Autologous or allogenic (latter better) but only 25% will have matched relative.
#50 Acquired
-Babies in nursery/day care have increased incidence of ALL
Inherited
-Fanconi anaemia- defect in DNA repair, majority get ca
-Downs- x20 risk of ALL
#53 May transform to high-grade lymphoma (Richter’s syndrome) a complication of B cell chronic lymphocytic leukemia (CLL) or hairy cell leukemia (HCL) in which the leukemia changes into a fast-growingdiffuse large B cell lymphoma. 5% of all CLL
Prolymphocytic transformation- increased numbers of circulating prolymphocytes, may be refractory to treatment.
#58 Usually seen on karyotyping but can also be observed on FISH if this is difficult. Ph chromosome occasionally seen in ALL (=bad!) and even more rarely AML.
#60 More banded (immature) neutrophils- left shift. Must be differentiated from leukmoid reaction (i.e. physiological reaction to stress, infection).
#64 Severe systemic symptoms include <10% weight loss, extreme fatigue, fever, night sweats
Monoclonal use still in early stages, different responses according to specific cytogenetics e.g. alemtuzumab in p53 mutations for clearing BM
BM transplantation in young patients, but delay until development of refractory disease worsens outcome.
#65 BM Tx may still be important in younger individuals or with HLA-identical siblings. Ideally in chronic phase.
#66 CLL- Monoclonal use still in early stages, different responses according to specific cytogenetics e.g. alemtuzumab in p53 mutations for clearing BM
BM transplantation in young patients, but delay until development of refractory disease worsens outcome.
CML- Role post-imatinib? In younger patients, ideally in chronic phase for up to 60% 10 year survival rates. BM Tx may still be important in younger individuals or with HLA-identical siblings. Ideally in chronic phase.
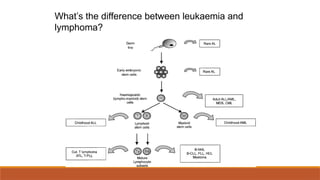
#77 The type of cell (how differentiated) they originate from. Can transform from one to the other- a continuum. But lymphoma usually initially populates LN, spleen etc. Lymphoma- LN origin, forming tumour mass
Leukaemia- BM origin, manifest in peripheral blood
But it’s not always easy to distinguish the two!
#80 AI conditions e.g. Sjorgren’s- non-H lymphoma- salivary extranodal marginal zone B cell lymphomas (MALT lymphomas in the salivary glands) and diffuse large B-cell lymphoma, increased in NHL generally in AI like RA, sarcoid, IBD,
#81 Consitutitive activation of NF-kB, role of EBV?
#82 Other symptoms:
Rash
-Cutaneous involvement, only as late complication
Abdominal pain, early satiety
-Splenomegaly but unusual as rarely massive
Shortness of breath, pleuritic chest pain, SVC syndrome
-Mediastinal involvement, pleural effusion, especially nodular sclerosing type
#83 Evidence of BM failure on bloods (e.g. anaemia, lymphopenia) is prognostic- bad!
Bx especially if elderly, advanced stage, systemic symptoms or high-risk histology (i.e. select stage II and above)
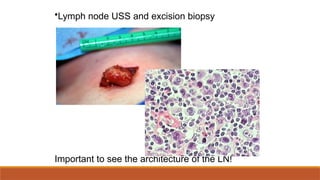
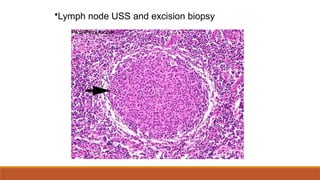
#84 Be sure it’s not carcinoma! Excision biopsy can promote spread. Core biopsy may be acceptable but important to examine architecture.
Mixed cellularity subtype-
Numerous R-S cells, mixed inflammatory background, obliteration of normal architecture
#87 Fertility e.g. sperm cyropreservation, embyro banking
Cardiac function- many agents cardiotoxic especially anthracyclins like doxorubicin
Respiratory function- bleomycin causes RPD
Allopurinol/uricase for tumour lysis syndrome
Others, as indicated (see leukaemias slide
#88 Typical chemo regimen ABVD
Adriamycin (doxorubicin/Hydroxydaunorubicin, the H in CHOP)
bleomycin
vinblastine
dacarbazine
Surgery not really used.
#90 Infection- direct transformation e.g. EBV, HTLV-1, HHV8 or chronic inflammation e.g. HCV, H pylori
#92 More varied than Hodgkin’s but LN and systemic symptoms still more important.
#93 Autoimmune (commoner in low grade) or BM infiltration e.g. anaemia
#94 Be sure it’s not carcinoma! Excision biopsy can promote spread. Core biopsy may be acceptable but important to examine architecture.
#95 Start with milder e.g. chlorambucil in low grade
Surgery can also be used for complications e.g. bulky splenomegaly etc.
Monoclonals can occasionally be used.
#96 Indolent lymphomas- curable if caught early but often not, don’t always respond well to chemo (monoclonals in follicular lymphoma). Relapse may occur years later.
Aggressive- symptomatic early on, may be curable with aggressive therapy but relapse often occurs soon after chemo e.g. 2y in diffuse large B cell lymphoma. May or may not be responsive to chemo. Most 5y survival patients cured.