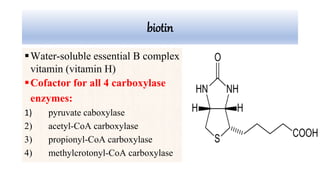
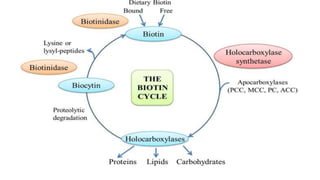
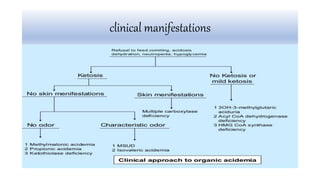
Multiple carboxylase deficiency is a recessive inherited amino acidopathy caused by deficiencies in holocarboxylase synthetase or biotinidase, enzymes necessary for biotin-dependent carboxylases. This leads to reduced activity of carboxylases and accumulation of toxic metabolites. Symptoms include metabolic acidosis, ketosis, hyperammonemia, hypoglycemia, and abnormal organic acids. Diagnosis is by newborn screening, enzyme assays, and urine organic acid analysis. Treatment involves biotin supplementation, which is effective if started early and can prevent symptoms and health problems if untreated.