CONTENTS
• INTRODUCTION
• NORMALLEUKOCYTE COUNT IN DIFFERENT AGE GROUP
• AETIOLOGY AND RISK FACTORS
• PATHOGENESIS
• CLASSIFICATION
• ACUTE MYELOID LEUKEMIA
• CHRONIC MYELOID LEUKEMIA
• ACUTE LYMPHOBLASTIC LEUKEMIA
• CHRONIC LYMPHOBLASTIC LEUKEMIA
• RECENT ADVANCES IN DIAGNOSIS OF LEUKEMIAS
3.
INTRODUCTION
• According toNational Library of Medicine,
• Leukemia is a heterogeneous group of hematologic malignancies that arise from the
dysfunctional proliferation of developing leukocytes.
• According to Robbins Basic Pathology 8th
Edition,
• These are the tumors that primarily involve the bone marrow, with spillage of neoplastic cells
into the peripheral blood.
4.
HISTORICAL PERSPECTIVE
• 1811,Peter Cullen defined a case of splenitis acutus with unexplainable milky blood
• Alfred Velpeau defined the leukemia associated symptoms, and observed pus in the blood
vessels (1825)
• John Bennett named the disease leucocythemia, based on the microscopic accumulation of
purulent leucocytes (1845).
• Rudolf Virchow defined a reversed white and red blood cell balance. He introduced the disease
as leukämie in 1847.
• Henry Fuller performed the first microscopic diagnose of a leukemic patient during life (1846).
Kampen KR. The discovery and early understanding of leukemia. Leuk Res. 2012 Jan;36(1):6-13
5.
TOTAL LEUCOCYTE COUNTACROSS DIFFERENT AGE GROUPS
• ADULTS - 4,000-11,000 per mm3 of blood
• AT BIRTH - 8,000-18,000 per mm3 of blood
• AT 1 year of age - 4,000-15,000 per mm3 of blood
• 6-12 years of age - 5,000-11,000 per mm3 of blood
• During Pregnancy - up to 15,000 per mm3 of blood
AETIOLOGY AND RISKFACTORS
• Chromosomal Abnormalities / changes
• Congenital Disorders
• Environmental factors
• Family History
• Chemical Agents
• Chemotherapeutic agents
• Viruses
9.
CLASSIFICATION
• In collaborationwith the Society for Hematopathology and the European
Association for Hematopathology , the World Health Organization (WHO)
published the third and fourth editions of the WHO Classification of Tumors of
Hematopoietic and Lymphoid Tissues, in 2001 and 2008, respectively, as part of a
series of WHO Classification of Tumors “blue book” monographs. The recent
changes in this classification has been done by WHO in 2022.
MYELODYSPLASTIC/MYELOPROLIFERATIVE NEOPLASMS.
chronic myelomonocyticleukaemia
Myelodysplastic/myeloproliferative neoplasm with neutrophilia
Myelodysplastic/myeloproliferative neoplasm with SF3B1 mutation and thrombocytosis
Myelodysplastic/myeloproliferative neoplasm, not otherwise specified
Khoury, J.D., Solary, E., Abla, O. et al. The 5th edition of the World Health Organization Classification of
Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia 36, 1703–1719 (2022).
12.
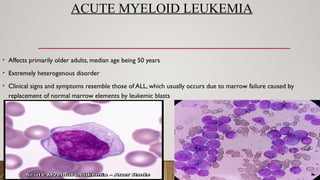
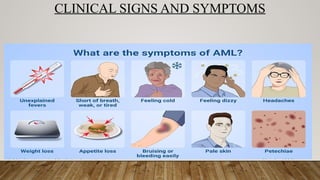
ACUTE MYELOID LEUKEMIA
•Affects primarily older adults, median age being 50 years
• Extremely heterogenous disorder
• Clinical signs and symptoms resemble those of ALL, which usually occurs due to marrow failure caused by
replacement of normal marrow elements by leukemic blasts
13.
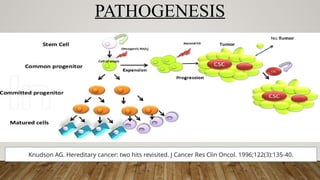
PATHOGENESIS
• Associated withacquired mutations in transcription factors that inhibit normal myeloid differentiation, leading to
accumulation of cells at earlier stages of development.
• Translocation t(15;17) fusion of RARA gene on chromosome 17 with PML gene on chromosome 15
• Produces fusion protein that block myeloid differentiation at the promyelocytic stage, probably by inhibiting the
normal function of RARA receptor.
• Also may occur as a result of gain-of-function mutations in FLT3.
MORPHOLOGIC CHARACTERISTICS
• Obtaininga peripheral blood smear is crucial for the examination. Characteristic features, in addition to
generalized thrombocytopenia, include blasts, which are large, immature leukocytes with a high nuclear-to-
cytoplasmic ratio, irregular nuclear contour, and smooth chromatin with prominent or multiple nucleoli. Blasts
have cytoplasm that appears pale or deep blue with a variably eosinophilic hue.
• Notably, a specific subtype of AML—acute promyelocytic leukaemia (APL)—exhibits a distinctive and
pathognomonic feature on peripheral blood morphology of abundant cytoplasmic Auer rods, which resemble
clumps of azurophilic granules elongated like needles.
Bain BJ, Béné MC. Morphological and Immunophenotypic Clues to the WHO Categories of Acute Myeloid Leukaemia. Acta
Haematol. 2019;141(4):232-244.
18.
HISTOCHEMISTRY
• Positive formyeloperoxidase
• Auer rods (linear azurophilic inclusions in the cytoplasm of blast cells) are intensely peroxidase positive,
pathognomic for AML.
• Monocytic differentiation demonstrated by staining with lysosomal nonspecific esterases.
• The diagnosis of acute myeloid leukemia requires equal to or greater than 20% blasts (except in
some cases with specific cytogenetic abnormalities or in erythroleukemia).
• The diagnostic work up typically includes morphologic, cytochemical, and immunophenotypic
features. This generally includes evaluation of the peripheral blood, bone marrow aspirate, and
bone marrow trephine biopsy
Cruise MW. Immunohistochemistry in Acute Myeloid Leukemia. Methods Mol Biol. 2017;1633:33-49
19.
IMMUNOPHENOTYPE
• Expression ofimmunologic markers is heterogenous.
• Most express combination of myeloid-associated antigens , such as CD13, CD14, CD15,CD64 or CD117 (cKIT) .
• CD33 is expressed on pluripotent stem cells, but is retained on myeloid progenitor cells.
20.
PROGNOSIS
• It isa devastating disease
• Karyotype aberrations (t[8;21], inv [16]) are associated with 50% chance of long term disease-free survival
• With conventional chemotherapy 15 -30% chances of survival
• Cytogenetic and molecular analyses provide the strongest prognostic information available, predicting outcome of
both remission induction and post remission therapy.
• Cytogenic and molecular information has been combined to form distinct prognostic groups
Döhner H, Estey E, Grimwade D, et al.: Diagnosis and management of AML in adults: 2017 ELN
recommendations from an international expert panel. Blood 129 (4): 424-447, 2017.
21.
CHRONIC MYELOGENOUS LEUKEMIA(CML)
• Principally affects adults between 26-60 years of age ; peak incidence is at fourth to fifth decade of life
• Accounts for 15-20% of all the cases of leukaemia
• Marked by hyperproliferation of neoplastic myeloid progenitors that retain the capacity for terminal
differentiation: resulting in increase in one or more formed elements of the peripheral blood.
• These tend to seed to secondary haematopoietic organs , resulting in hepatosplenomegaly and lymphadenopathy.
22.
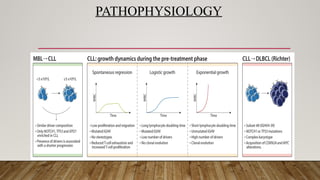
PATHOPHYSIOLOGY
Faderl S, TalpazM, Estrov Z, O'Brien S, Kurzrock R, Kantarjian HM. The biology of chronic myeloid leukemia. N
Engl J Med. 1999 Jul 15;341(3):164-72
CLINICAL FEATURES
• Onsetis slow, with nonspecific symptoms
• Sometimes, the first symptom is a dragging sensation in the stomach , due to splenomegaly
• In more than half of the patients, the spleen size extends beyond 5 cm below the left costal margin at the time of
diagnosis
• Presence of Ph chromosome is the most definite way to differentiate it from leukemoid reactions
26.
MORPHOLOGIC CHARACTERISTICS
• Leukocytecount is elevated , often exceeding 100,000 cells/ microlitre
• Circulating cells are predominantly neutrophils, metamyelocytes and myelocytes but basophils and eosinophils are
also seen
• A small proportion of myeloblasts usually <5% is seen in PBS, Thrombocytosis is also typically seen
• Bone marrow is hypercellular
• Red pulp of spleen is seen resembling a bone marrow, because of extensive haematopoiesis
27.
HISTOCHEMISTRY
• Negative forLeukocyte Alkaline Phosphatase, which is positive in leukemoid reactions
• Negative for Myeloperoxidase, which can help in differentiating from AML
• Pseudo-Gaucher cells are histiocytes with rounded, blue, lamellar cytoplasm resembling "onion
skin" that can be found in up to 40% of the bone marrow of patients with CML. These are similar
to glucocerebroside-stuffed histiocytes seen in Gaucher disease.
Gören Şahin D, Üsküdar Teke H, Karagülle M, Andıç N, Gündüz E, Işıksoy S, Balić M, Akay OM. Gaucher Cells
or Pseudo-Gaucher Cells: That's the Question. Turk J Haematol. 2014 Dec 5;31(4):428-9.
28.
HISTOPATHOLOGY OF DIFFERENTPHASES OF CML
• Chronic Phase
• The peripheral blood smear will show a leukocytosis due to granulocytes in various stages of maturation. A bimodal
distribution with higher proportions of mature segmented neutrophils and myelocytes is seen with blast cells less than
2% of the white blood cells.
• Accelerated Phase
• The peripheral smear may or may not show increased blasts (10% to 19%). The bone marrow aspirate and biopsy will
show similar changes to chronic phase CML with increased blasts (10% to 19%).
• Blast Phase
• The peripheral smear and/or bone marrow aspirate will show greater than 20% blasts, or there will be an extramedullary
proliferation of blasts. Most cases will show blasts with myeloid differentiation
Cortes JE, Talpaz M, O'Brien S, Faderl S, Garcia-Manero G, Ferrajoli A, Verstovsek S, Rios MB, Shan J, Kantarjian HM.
Staging of chronic myeloid leukemia in the imatinib era: an evaluation of the World Health Organization proposal. Cancer. 2006
29.
IMMUNOPHENOTYPING
• BCR-ABL fusionprotein transcripts are seen in cytogenetic analysis
• Positive for CD34, CD117, CD3, UCHL-1, CD13, CD33, CD 10, TdT.
• Also positive for CD3+, CD4+ T-helper cells as well as CD45RO+ memory T cells
Thiele J, Kvasnicka HM, Titius BR, Parpert U, Nebel R, Zankovich R, Dienemann D, Stein H, Diehl V, Fischer R.
Histological features of prognostic significance in CML--an immunohistochemical and morphometric study
(multivariate regression analysis) on trephine biopsies of the bone marrow
30.
PROGNOSIS
• Course isslow progressive
• Without treatment, median survival rate is 3 years
• After a variable and unpredictable period, 50% of the patients enter into accelerated phase, which reduces their
response to treatment and also the survival chance
• This results into the final transformation resembling acute leukaemia (i.e.) Blast crisis
31.
ACUTE LYMPHOBLASTIC LEUKEMIA
•Acute lymphocytic leukemia (ALL) is a malignancy of B or T lymphoblasts characterized by uncontrolled
proliferation of abnormal, immature lymphocytes and their progenitors which ultimately leads to the replacement of
bone marrow elements and other lymphoid organs resulting in a characteristic disease pattern.
• Occurs predominantly in children and young adults
• Constitute 80% of all the childhood leukemia, peaking at 4 years of age
• Pre-T cell tumors are most common in adolescent males of between 15 and 20 years of age
32.
PATHOPHYSIOLOGY
• Principle pathogeneticmechanism- Block in Differentiation
• Accumulation of immature leukemic blasts in the marrow
• Supresses the normal haematopoietic function
• Eventually leading to bone marrow failure
33.
CLINICAL FEATURES
• Anemia,thrombocytopenia, and neutropenia due to the replacement of the bone marrow with the tumor.
• Symptoms can include fatigue, easy or spontaneous bruising and/or bleeding, and infections.
• Additionally, B-symptoms, such as fever, night sweats, and unintentional weight loss, are often present but may be
mild, and hepatomegaly, splenomegaly, and lymphadenopathy can be seen in up to half of adults on presentation.
• Central nervous system (CNS) involvement is common and can be accompanied by cranial neuropathies or
symptoms, predominantly meningeal, related to increased intracranial pressure.
34.
MORPHOLOGIC CHARACTERISTICS
• Blastscomprise >25% of the marrow cellularity, WBC count variable > or < 100,000 cells/microlitre
• Nuclei in Wright-Giemsa-stained preparations are somewhat coarse and clumped chromatin and one to two
nucleoli with anaemia and platelet count below 100,000cells/microlitre
• Cytoplasm of lymphoblasts often contain large aggregates of Periodic Acid – Schiff –positive material.
• Neutropenia is a common finding in PBS, sometimes may also show pancytopenia but no blasts in smear.
35.
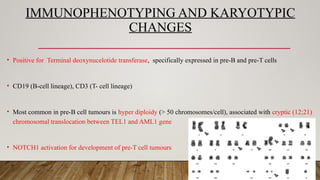
IMMUNOPHENOTYPING AND KARYOTYPIC
CHANGES
•Positive for Terminal deoxynucelotide transferase, specifically expressed in pre-B and pre-T cells
• CD19 (B-cell lineage), CD3 (T- cell lineage)
• Most common in pre-B cell tumours is hyper diploidy (> 50 chromosomes/cell), associated with cryptic (12;21)
chromosomal translocation between TEL1 and AML1 gene
• NOTCH1 activation for development of pre-T cell tumours
36.
PROGNOSIS
• Children of2 to 10 years of age have the best prognosis ; can be treated successfully
• Worse outcomes are associated with : male gender, age younger than 2 or older than 10 years of age and a high
leukocyte count at the time of diagnosis
• Tumours with rearrangements of MLL or Ph are associated with poor outcomes
• Good prognosis is associated with chromosomal aberrations in age group of 2-10 years of age
37.
CHRONIC LYMPHOBLASTIC LEUKEMIA
•Most common leukaemia of adults in the western world
• Much less common in Asia
• Resembles morphologically, phenotypically and genotypically with Small Lymphocytic Lymphoma
• affects both the peripheral blood and the bone marrow.
38.
CLINICAL FEATURES
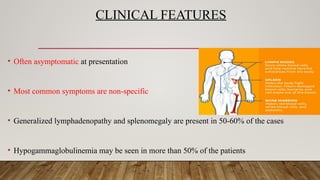
• Oftenasymptomatic at presentation
• Most common symptoms are non-specific
• Generalized lymphadenopathy and splenomegaly are present in 50-60% of the cases
• Hypogammaglobulinemia may be seen in more than 50% of the patients
MORPHOLOGICAL CHARACTERISTICS
• Sheetsof small round lymphocytes and scattered ill-defined foci of larger, actively dividing cells efface the
involved lymph node
• Cells are compact , small ,resting lymphocytes with dark-staining round nuclei, scanty cytoplasm and little
variation in size
• Foci of mitotically active centres proliferation centres Patho gnomic for ALL
• Bone marrow, spleen and liver are also involved in almost all of the cases
41.
IMMUNOPHENOTYPE, KARYOTYPING
• Positivefor pan-B cell markers - CD19 , CD20, CD23 surface immunoglobulin heavy and light chain
• Also express CD5 , which is also seen in mantle cell lymphoma
• 50% of patients have karyotyping abnormalities, most common being trisomy 12 and deletion of chromosome 11 and 12
• Chromosomal translocations are rare
• Most of them undergo somatic hypermutations of their immunoglobulin segments, consistent with memory B- cell
42.
DENTAL IMPLICATIONS INLEUKEMIA
• Petechiae, spontaneous bleeding and pain, mucosal ulceration, gingival hyperplasia and mucosal pallor. In some
cases, dysfunction of salivary glands and temporomandibular joint is observed. Neurotoxicity, mucosal fibrosis,
radiation osteonecrosis, or soft tissue necrosis after the treatment ends. Patients have impaired healing due to
reduced anti-inflammatory defenses and are more susceptible to viral opportunistic infections, secondary bacterial
infections, or candidiasis
• Biofilm accumulation and poor oral hygiene are known major risk factors for gingivitis or periodontitis,
especially in patients with systemic diseases, including leukemia
Padmini C., Bai K.Y. Oral and Dental Considerations in Pediatric Leukemic Patient. ISRN Hematol. 2014;2014:895721.
43.
RECENT ADVANCES INDIAGNOSIS OF LEUKEMIAS
• With the development of new technologies in the diagnosis of leukemia, modification has been
towards the method of approaching individual cases.
• In addition to morphological examination, cytochemistry, immunophenotyping, electron
microscopy and cytogenetics are now routinely used for daily practice.
• Flow cytometry and FISH can be used for evaluating the Ph chromosome as well as in CLL
44.
REFERENCES
• Robbins basicpathology,8th
Edition
• Kampen KR. The discovery and early understanding of leukemia. Leuk Res. 2012 Jan;36(1):6-13
• Knudson AG. Hereditary cancer: two hits revisited. J Cancer Res Clin Oncol. 1996;122(3):135-40
• Khoury, J.D., Solary, E., Abla, O. et al. The 5th edition of the World Health Organization Classification of
Hematolymphoid Tumors: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia 36, 1703–1719 (2022)
• Bain BJ, Béné MC. Morphological and Immunophenotypic Clues to the WHO Categories of Acute Myeloid
Leukaemia. Acta Haematol. 2019;141(4):232-244
• Döhner H, Estey E, Grimwade D, et al.: Diagnosis and management of AML in adults: 2017 ELN
recommendations from an international expert panel. Blood 129 (4): 424-447, 2017














![PROGNOSIS
• It is a devastating disease
• Karyotype aberrations (t[8;21], inv [16]) are associated with 50% chance of long term disease-free survival
• With conventional chemotherapy 15 -30% chances of survival
• Cytogenetic and molecular analyses provide the strongest prognostic information available, predicting outcome of
both remission induction and post remission therapy.
• Cytogenic and molecular information has been combined to form distinct prognostic groups
Döhner H, Estey E, Grimwade D, et al.: Diagnosis and management of AML in adults: 2017 ELN
recommendations from an international expert panel. Blood 129 (4): 424-447, 2017.](https://image.slidesharecdn.com/leukemia-250912111543-f4d90b24/85/LEUKEMIA-BASICS-AND-THE-CHARACTERISTICS-20-320.jpg)