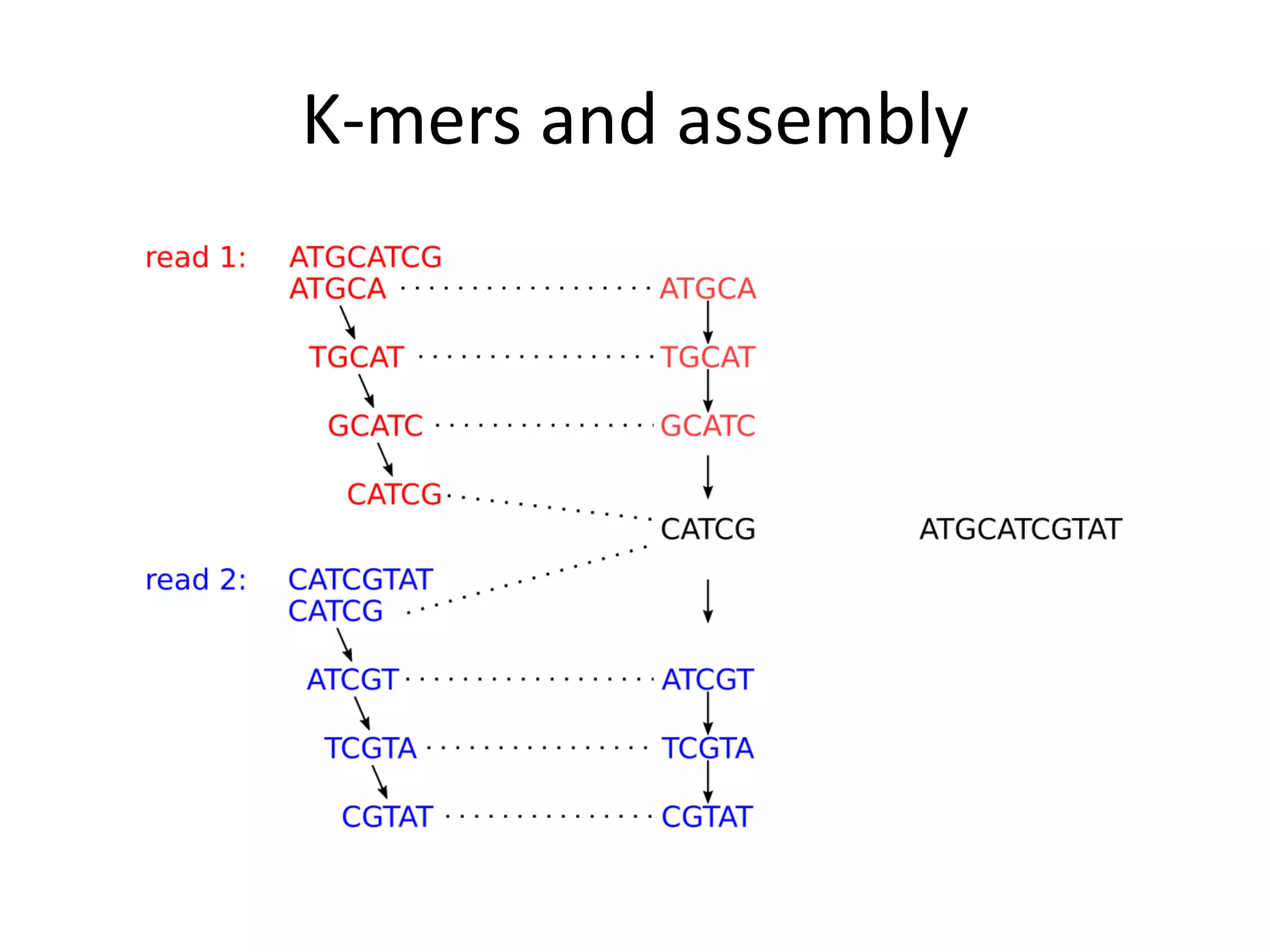
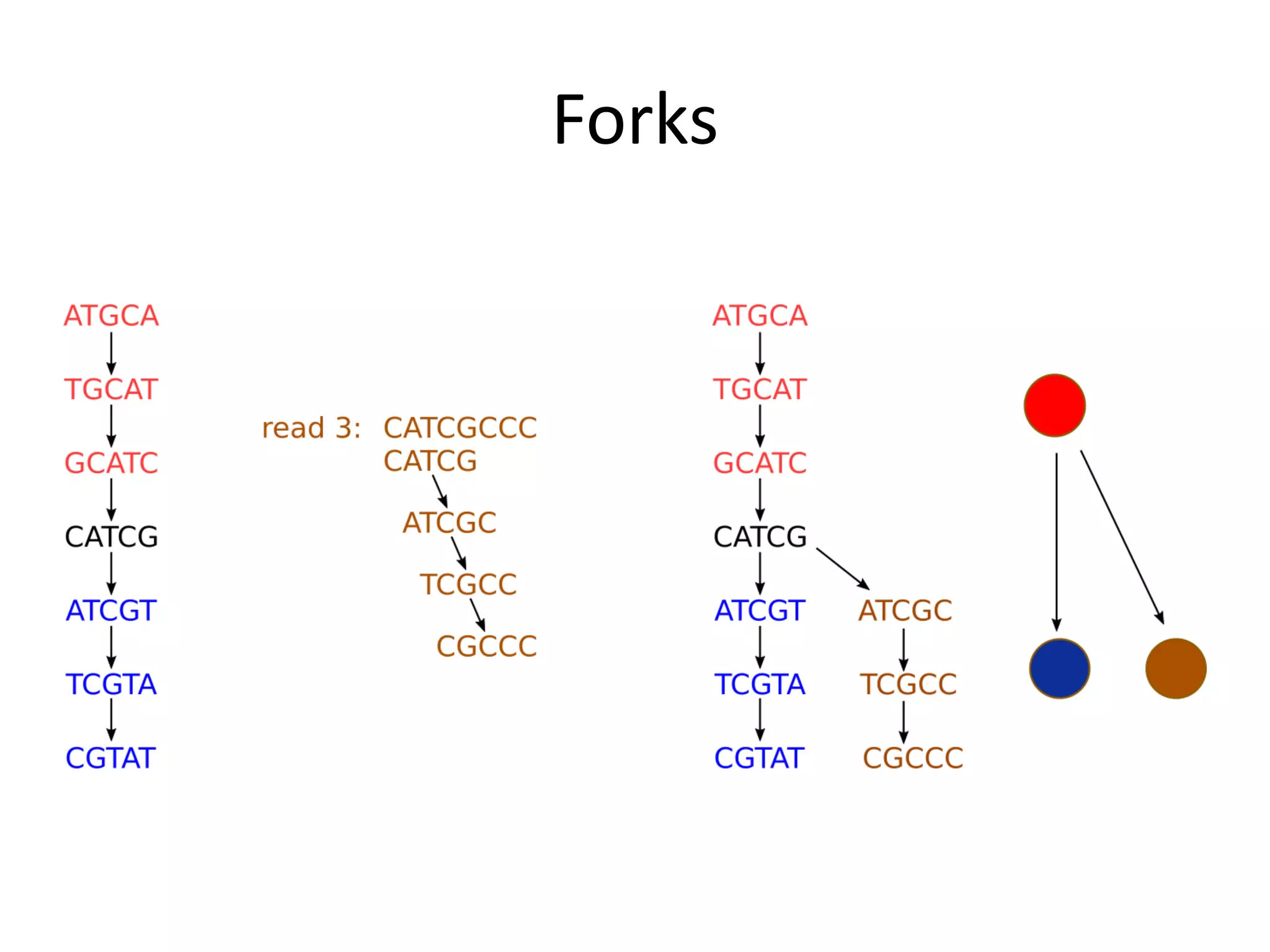
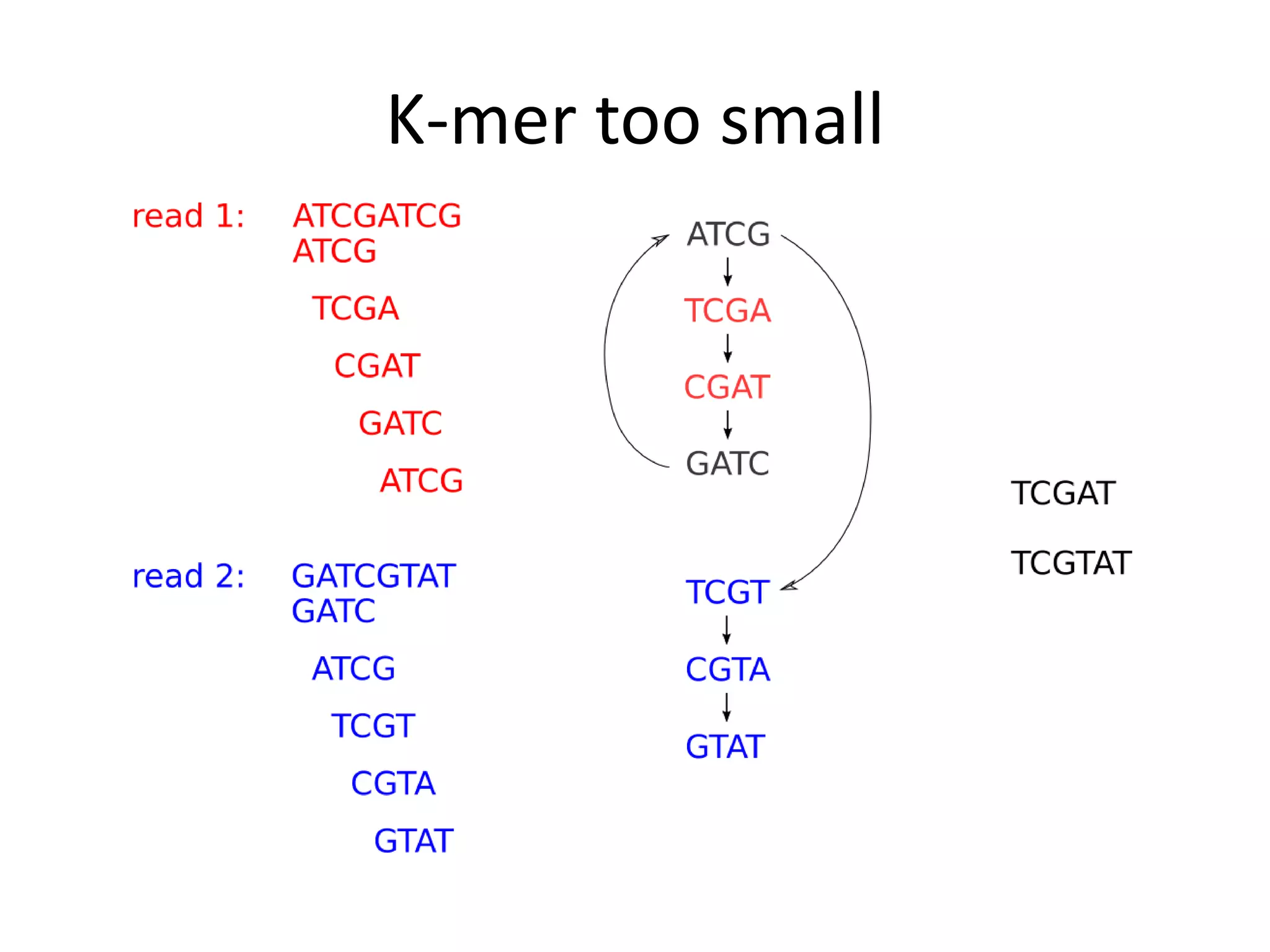
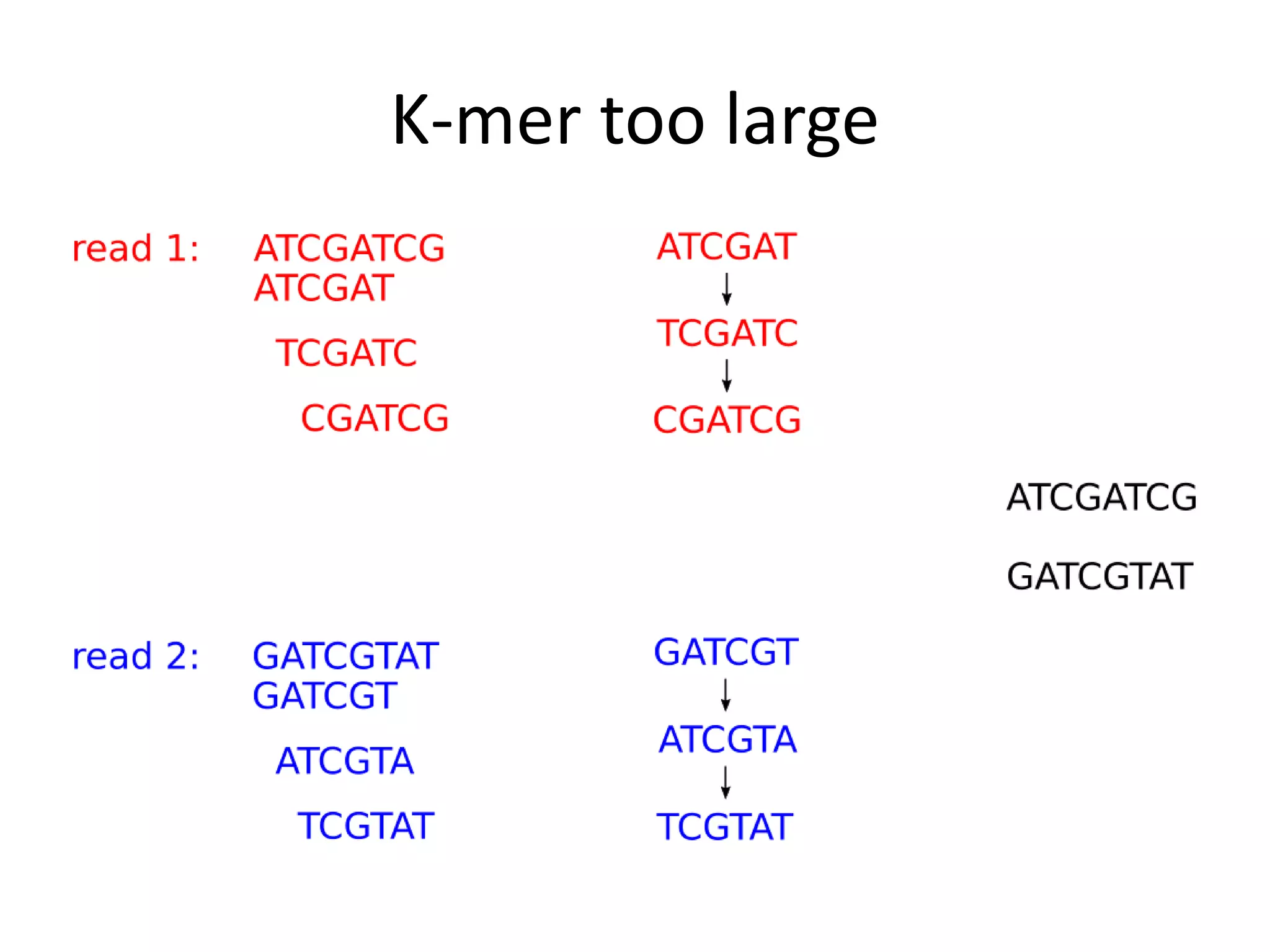
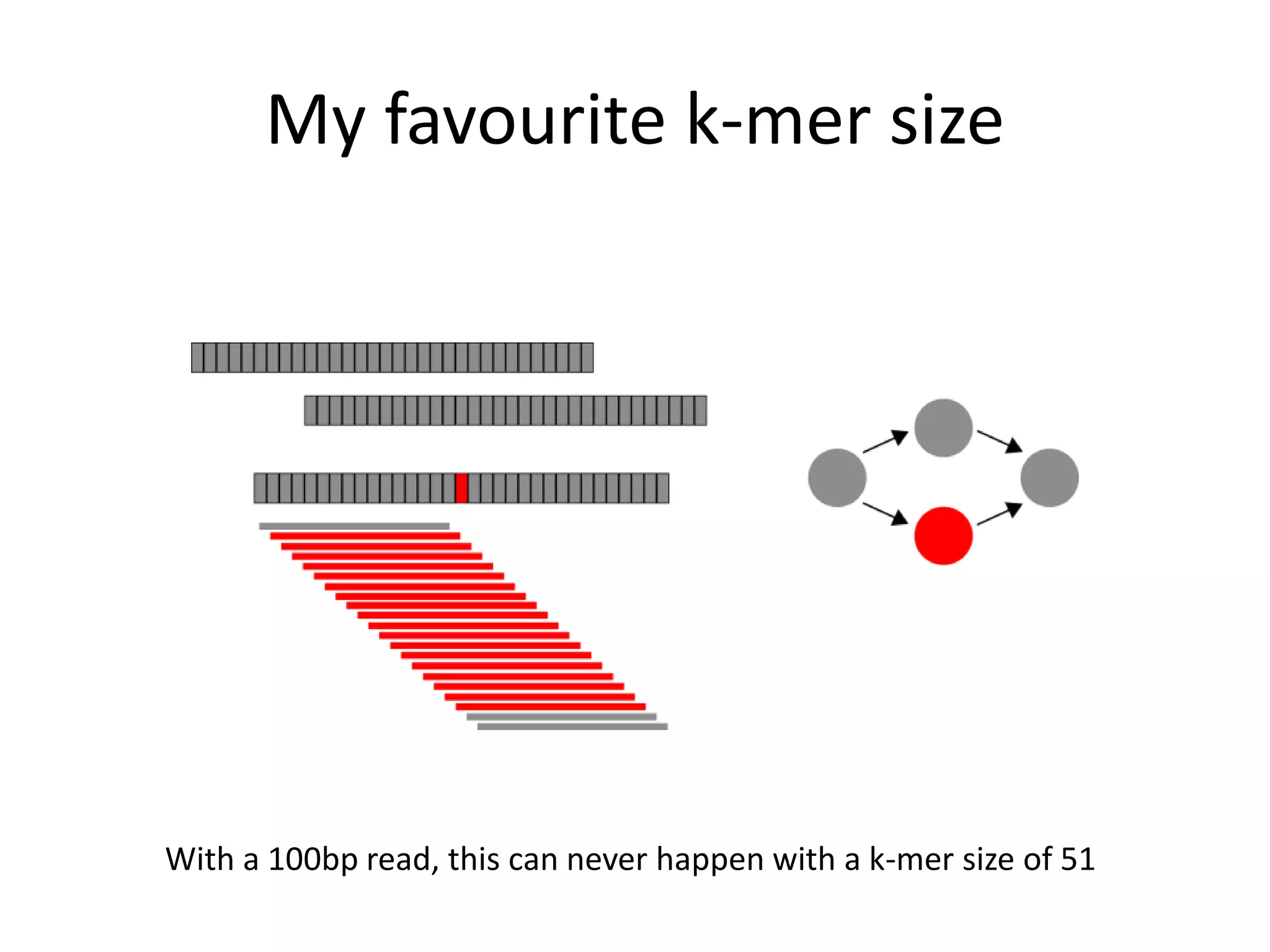
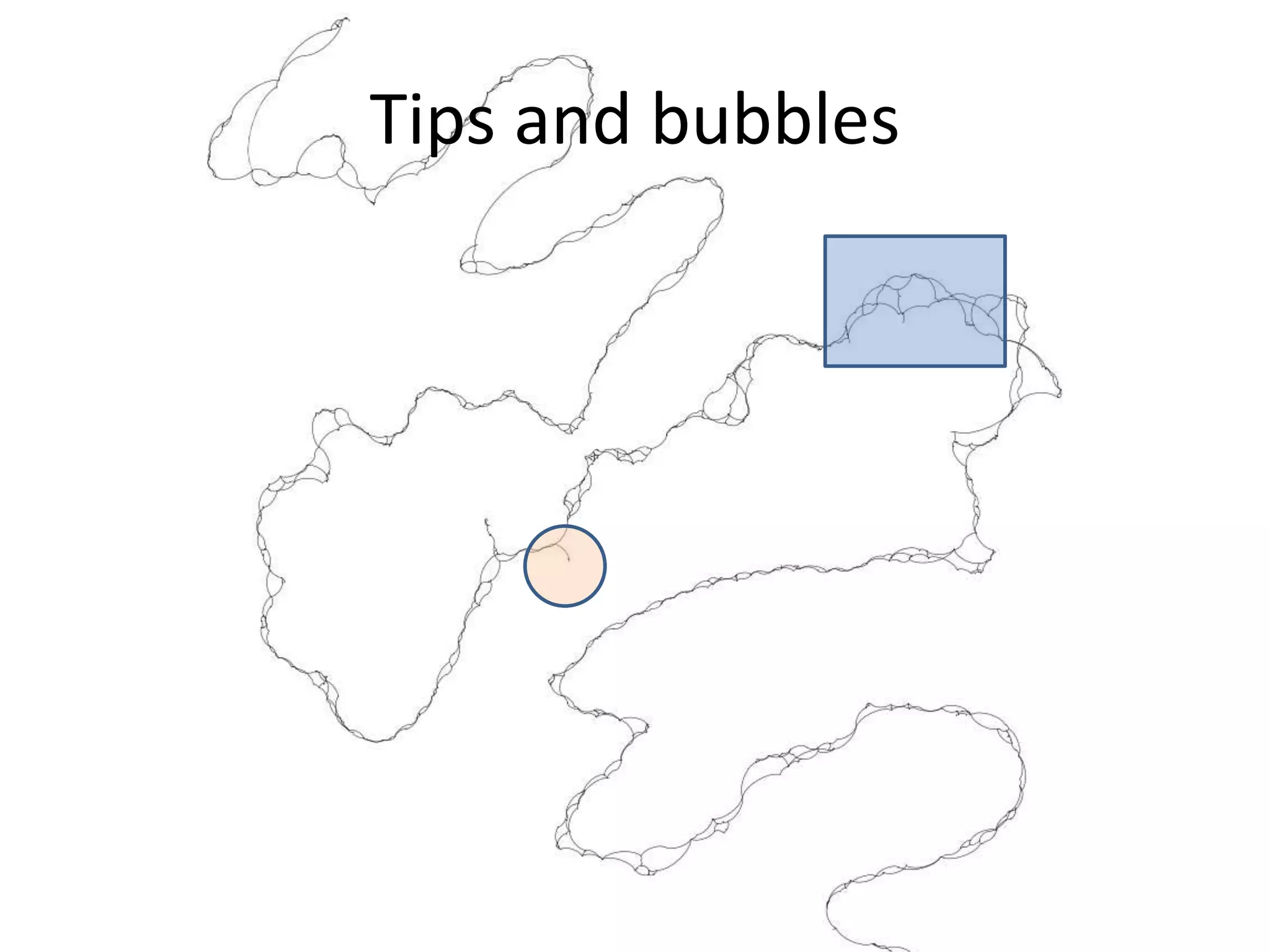
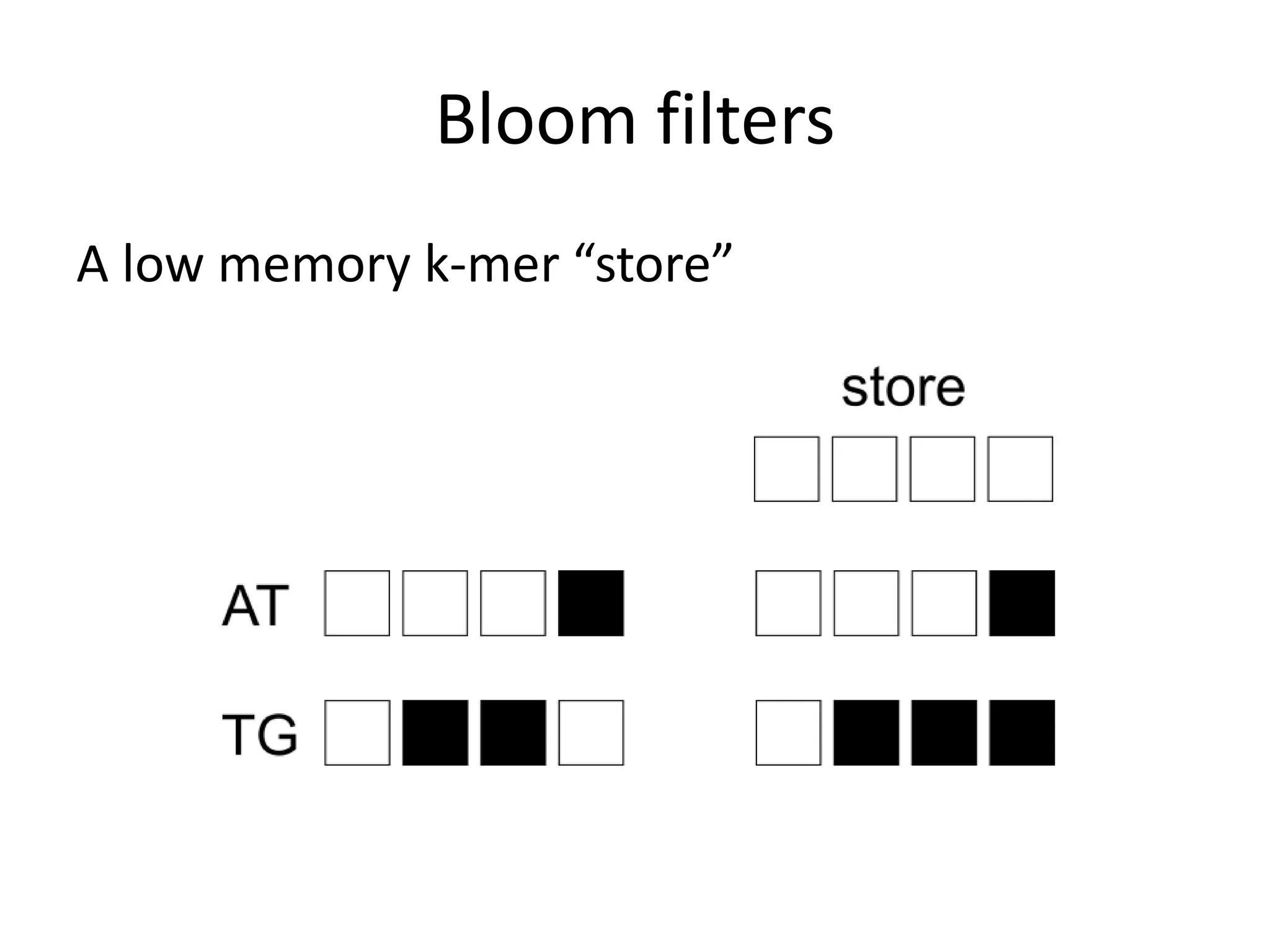
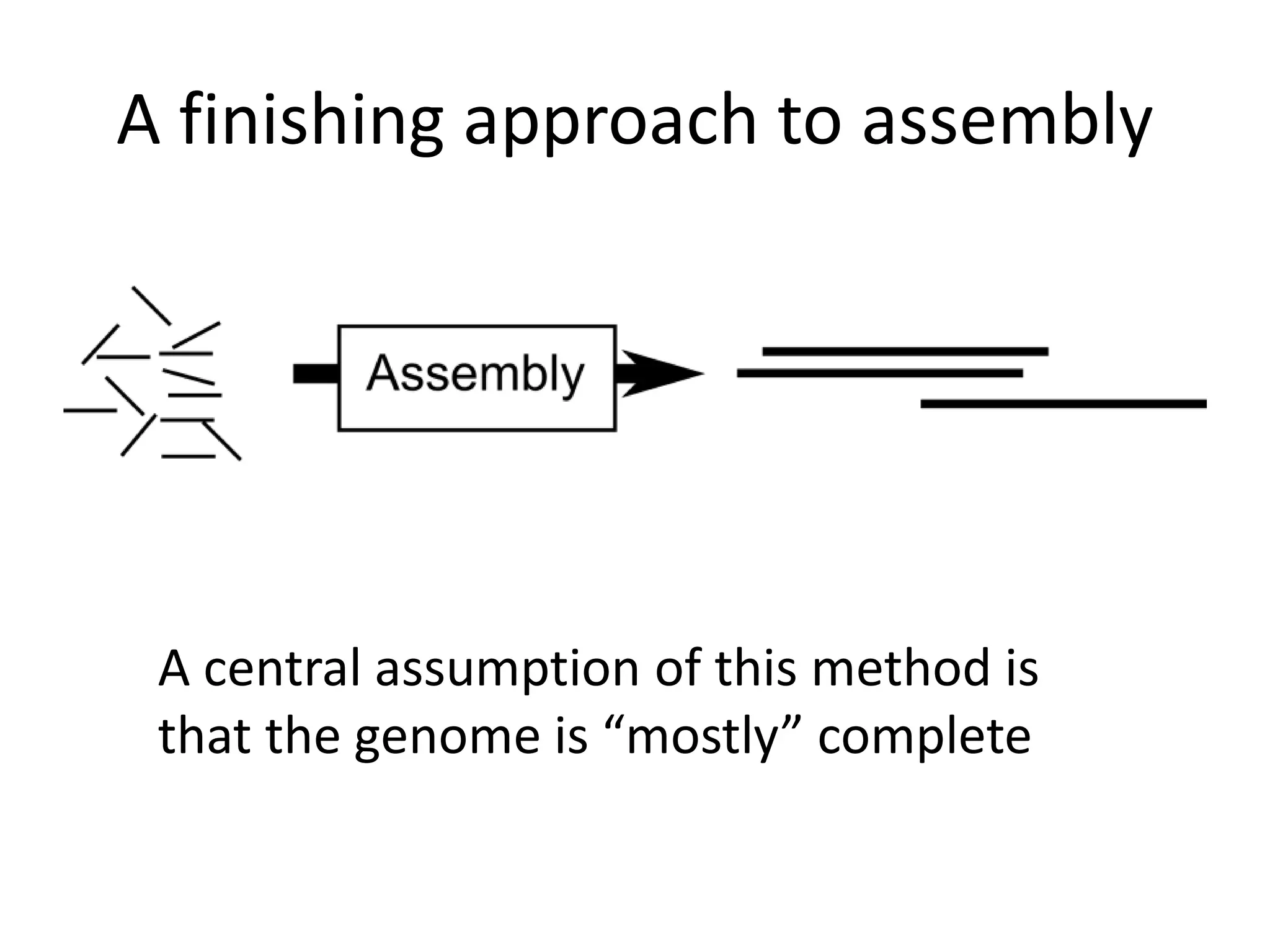
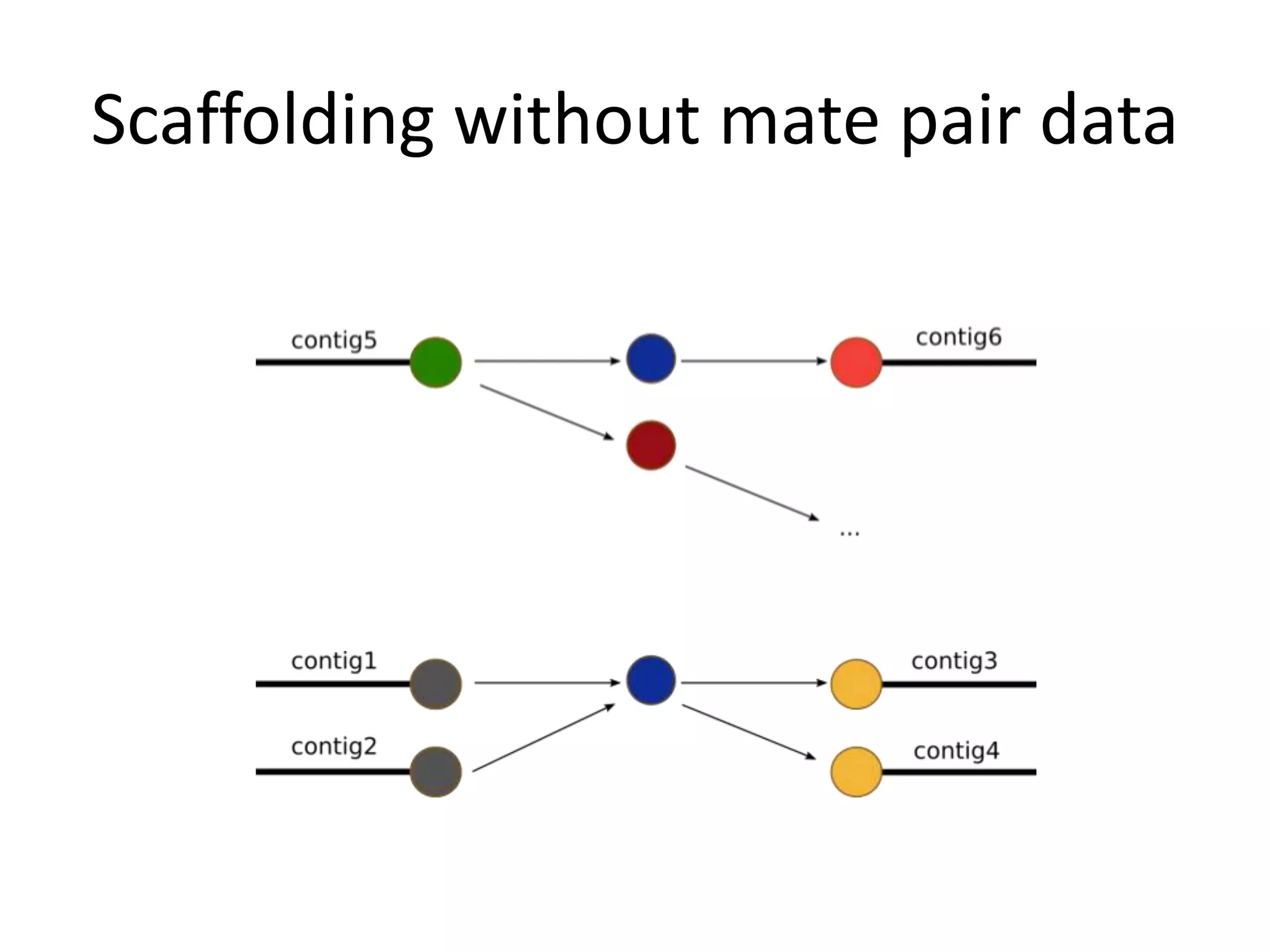
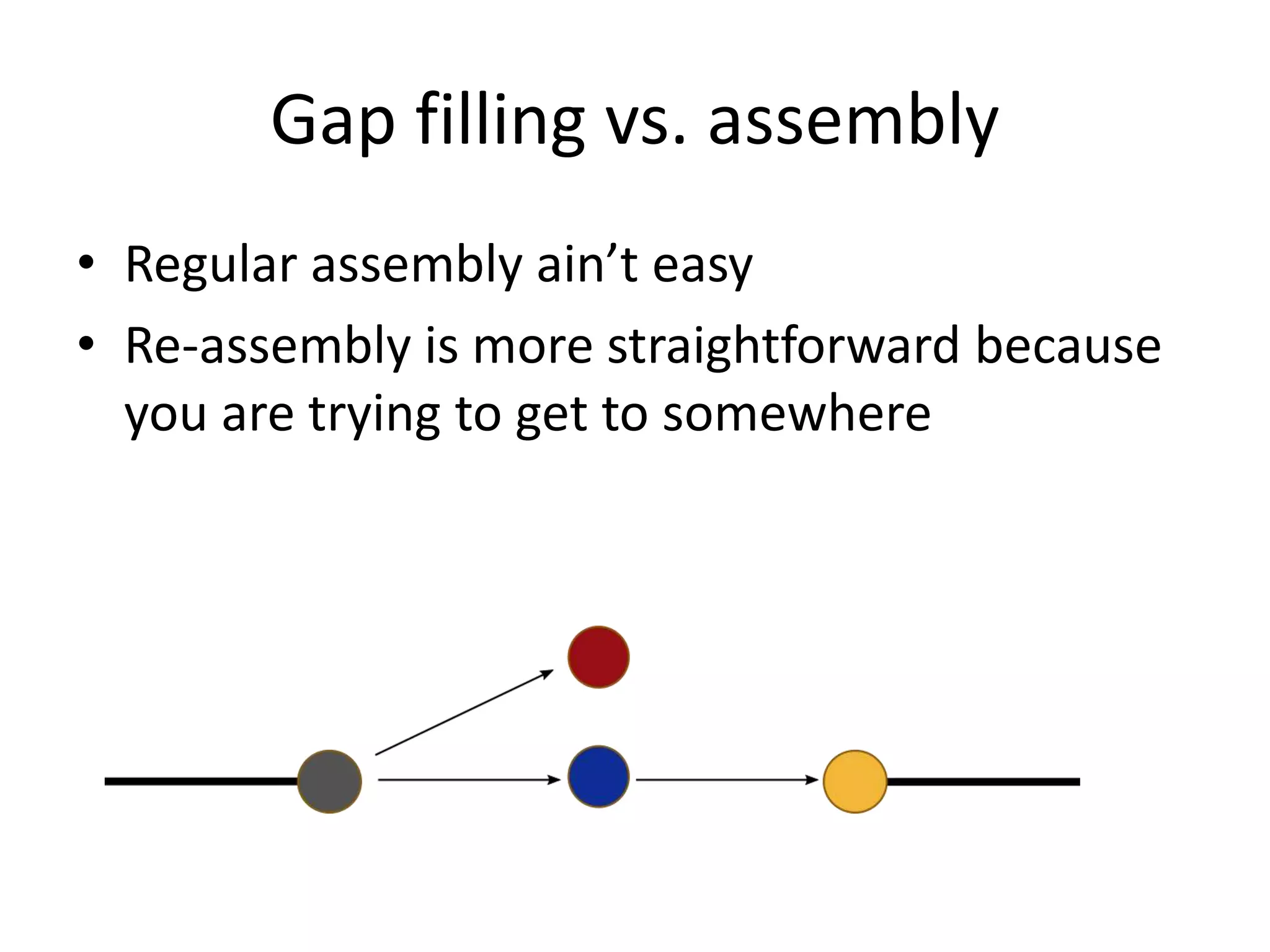
This document summarizes a presentation about using de Bruijn graphs for genome assembly from short read sequencing data. It discusses how de Bruijn graphs make assembly of millions of reads tractable, and addresses challenges like choosing the right k-mer size, handling tips and bubbles, and dealing with memory issues in metagenome assembly. It also presents solutions to memory problems, describes how lossy de Bruijn graphs work, and explains how gap filling can correct assembly errors and account for strain variation.















































![Coded Agents – with UiPath SDK + LangGraph [Virtual Hands-on Workshop]](https://cdn.slidesharecdn.com/ss_thumbnails/codedagentsdeck-251215155422-5497c599-thumbnail.jpg?width=640&height=640&fit=bounds)









