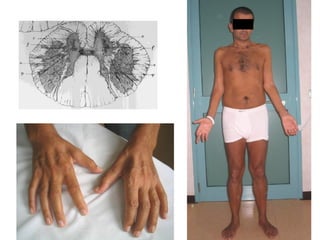
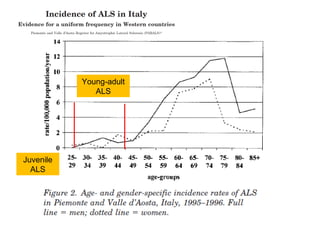
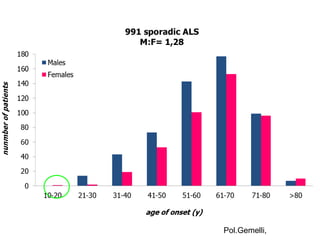
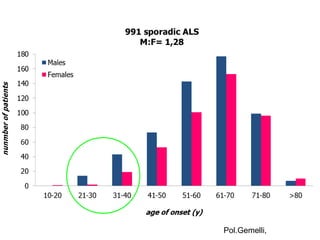
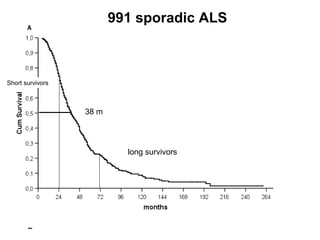
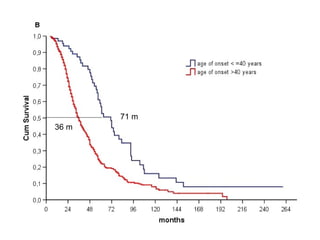
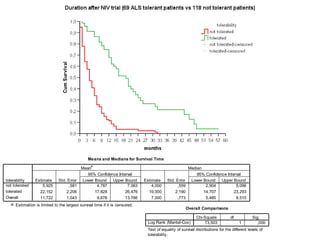
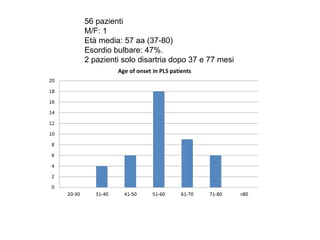
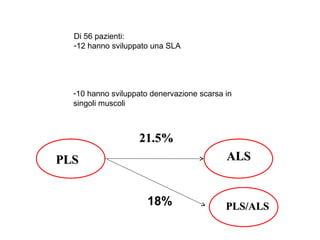
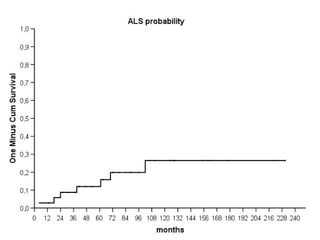
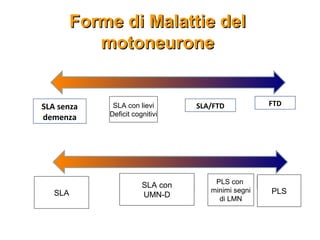
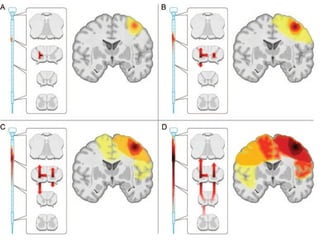
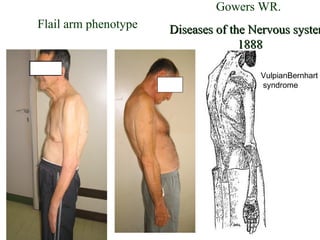
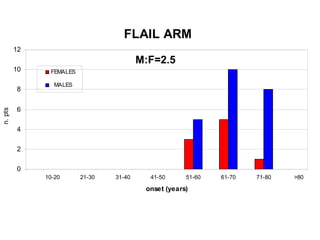
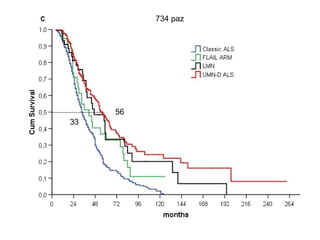
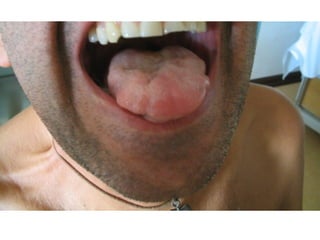
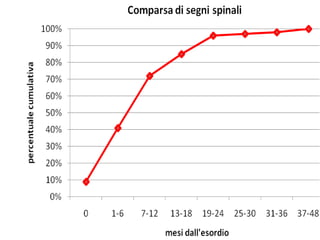
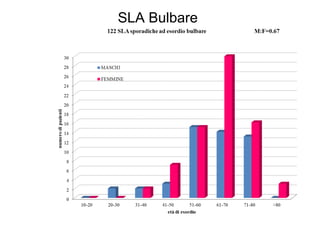
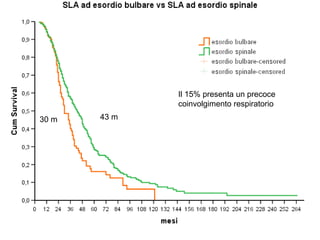
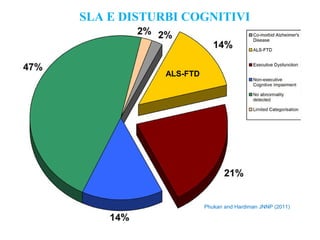
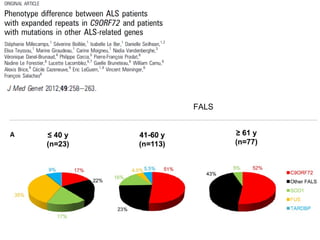
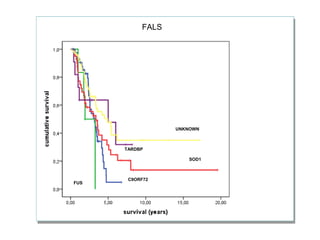
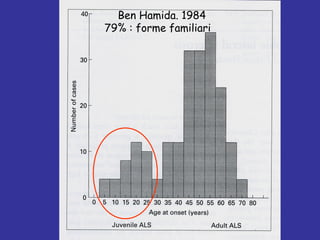
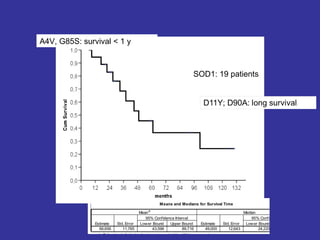
Il documento discute l'eterogeneità fenotipica della sclerosi laterale amiotrofica (SLA), evidenziando variabili come l'età di esordio, la durata della malattia e le condizioni associate come FTD e Parkinson. Viene analizzata la SLA giovanile e l'ALS di esordio giovane-adulto, sottolineando differenze nel quadro clinico e nella sopravvivenza. Inoltre, si menzionano vari fenotipi di malattia e la presenza di segni clinici e elettrofisiologici peculiari.