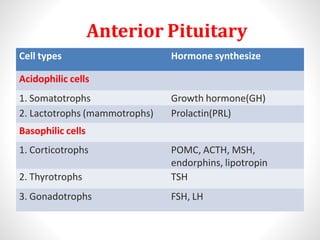
Hypopituitarism is a condition caused by insufficient production of hormones by the pituitary gland. The pituitary gland is divided into the anterior and posterior pituitary. The anterior pituitary contains cell types that synthesize important hormones such as growth hormone, prolactin, ACTH, TSH, and gonadotropins. Hypopituitarism can be caused by developmental defects, tumors, vascular issues, trauma, infections, or autoimmune disorders. Common symptoms depend on which hormones are deficient but may include short stature, weight changes, hypoglycemia, fatigue, and sexual dysfunction. Several genetic syndromes like septo-optic dysplasia, Prader-Willi syndrome, and Kallman syndrome can also cause