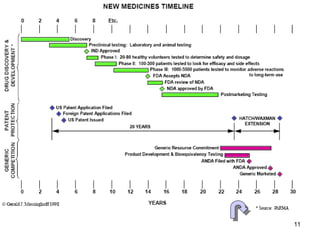
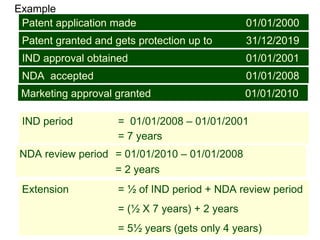
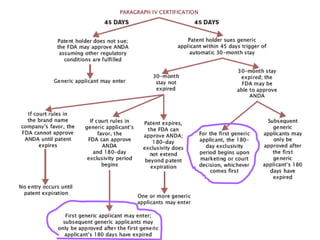
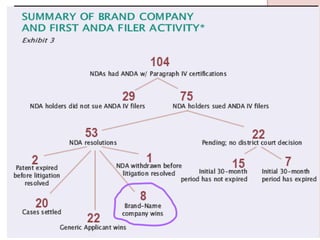
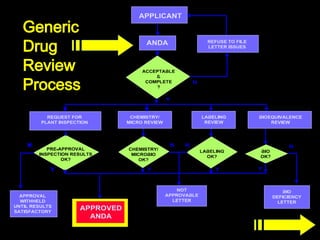
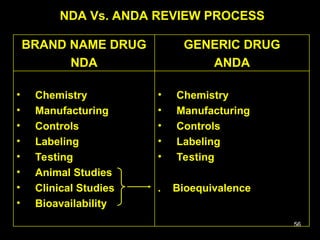
The document discusses the impact of the Hatch-Waxman Act on India's pharmaceutical industry, highlighting how it has facilitated growth through changes in patent regulations and increased access to international markets. It details the act's objectives, such as reducing drug prices and encouraging research, while describing the complexities involved in obtaining drug approvals via abbreviated new drug applications (ANDAs) and the challenges associated with patent litigation. Furthermore, it outlines various strategies used by companies to navigate the regulatory landscape, including para IV filings and loopholes exploited within the patent system.










































![47
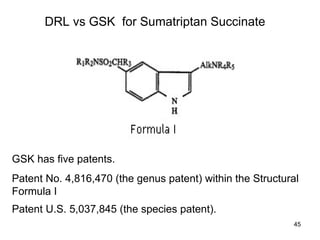
• U.S. Patent # 5,863,559 for pharmaceutical composition
for film-coated tablet of 3-[2-(dimethylamino)ethyl]-N-
methyl-1H-indole-5-methanesulphonamide succinate
(1:1) salt as active ingredient.
• U.S. Patent # 6,020,001 for pharmaceutical composition
for film-coated tablet of 3-[1-(dimethylamino)ethyl]-N-
methyl-1H-indole-5-methanesulphonamide succinate
(1:1) salt as active ingredient.
• U.S. Patent # 6,368,627 directed to a method of treating
or prophylactically treating a human suffering from
migraine which comprises oral administration of a
pharmaceutical composition comprising a film-coated
solid dosage form of 3-[2-dimethylamino)ethyl]-N-methyl-
1H-indole-5-methanesulfonamide or a pharmaceutically
acceptable salt or solvate therefore as active ingredient.
DRL vs GSK for Sumatriptan Succinate](https://image.slidesharecdn.com/hatchwaxmanact-250201043037-133ed248/85/Hatch-Waxman-Act-for-Generic-ANDA-application-47-320.jpg)




![52
DRL’s unique way
• Pfizer Inc. had patents for amlodipine salts including
maleate and besylate
• Pfizer investigated the maleate salt all the way through
Phase III and then switched to the besylate salt, due to
stability problem with maleate.
• DRL attacked active ingredient patent for amlodipine
besylate in a unique way
• It filed NDA and not ANDA through section 505(b)(2)
[ similar to Paper NDA] using the clinical trial data
submitted by Pfizer to market amlodipin maleate.
• Lower court gave verdict in favour of DRL
• Pfizer went for appeal](https://image.slidesharecdn.com/hatchwaxmanact-250201043037-133ed248/85/Hatch-Waxman-Act-for-Generic-ANDA-application-52-320.jpg)






















![Abbreviated New Drug Application [ANDA]](https://cdn.slidesharecdn.com/ss_thumbnails/abbreviatednewdrugapplicationanda-160619062810-thumbnail.jpg?width=640&height=640&fit=bounds)






























