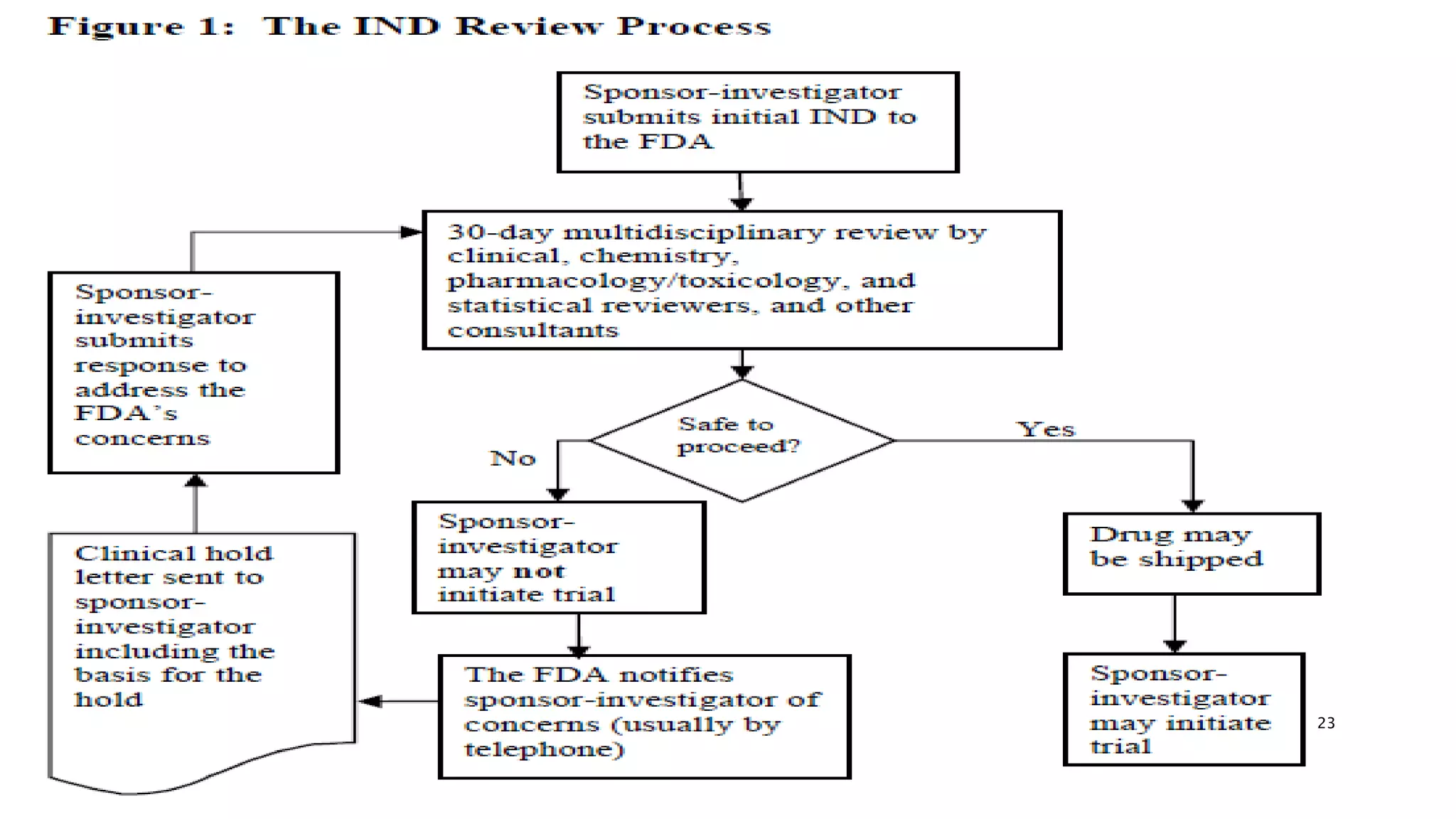
This document provides guidance on submitting an Investigational New Drug (IND) application to the FDA to request permission to begin human testing. It discusses the different types of INDs, including commercial, research, emergency use, and more. The document outlines the key components of an IND application, including preclinical data, manufacturing information, clinical protocols, regulations that apply, and ongoing reporting requirements like annual reports. It also discusses the IND review process, including clinical holds, amendments, and withdrawal of an IND. The overall purpose is to ensure the safety of human subjects and that the investigational product is reasonably safe to begin initial human testing.






![Regulations apply to the IND application process
21CFR Part 312 Investigational New Drug Application
21CFR Part 314 INDA and NDAApplications for FDAApproval to
Market a New Drug (New Drug Approval)
21CFR Part 316 Orphan Drugs
21CFR Part 58 Good Lab Practice for Nonclinical Laboratory
[Animal] Studies
21CFR Part 50 Protection of Human Subjects
21CFR Part 56 Institutional Review Boards
21CFR Part 201 Drug Labeling
21CFR Part 54 Financial Disclosure by Clinical Investigators
7](https://image.slidesharecdn.com/17mph202guidanceforindustryforindapplication-180416175848/75/Guidance-for-industry-for-IND-application-7-2048.jpg)























![establishing_pv_centers_in_industry_AND_NATIONAL_PROGRAMME[1].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/establishingpvcentersinindustryandnationalprogramme1-230725101256-d16cc241-thumbnail.jpg?width=640&height=640&fit=bounds)






























































