XERODERMA PIGMENTOSUM
• Dry,pigmented skin
• AR
• 1 in 10,000-30,000
• Genes involved- XPA-XPG and XPV variant (8 subtypes of XP).
• Defect is in NER
• GG-NER pathway involves XPC, XPE
• TC-NER pathway involves CSA, CSB
• Core NER pathway involves XPA, B, D, F, G genes
4.
➢ Clinical features:
•Normal at birth
• The development and severity of features depends upon the
amount of sun exposure and protection from UVR.
• Cutaneous manifestations start developing after 6 months of
age.
• Erythema, scaling and ephelides develop over sun exposed sites.
• XPC, XPE, XPV gene defects- no sunburn
- highest risk of skin cancers
- diagnosed late
- unique ocular sensitivity with XPC
5.
• CSA, CSBinvolvement- severe sunburnreactions
- hence, diagnosed early
- low risk of skin cancers
- neurological manifestations
(hyperreflexia, demyelination
, microcephaly)
• XPA, B, D, F, G gene involvement – both photosensitivity with
skin cancers and neurological manifestations (severe with XPA)
• De Sanctis Cacchione syndrome- rare variant of XP, presents
with severe neurologicalmanifestations, deafness, ataxia,
paralysis.
6.
• Ophthal- photophobia,ectropion, conjunctivitis, symblepheron
• Carcinoma- 10,000 fold risk of NMSC, 2000 fold risk of
melanoma.
• Death is mostly due to metastatic melanoma or invasive SCC by
29-32 years of age.
• Median age for dev. of NMSC-9 years, for melanoma- 22 years.
➢ Investigations:
• Prenatal diagnosis
• Postnatal- fibroblastsare irradiated with UVR, UDS is measured
• Decreased level of UDS indicates XP
• XPV- irradiated cells are incubated with caffeine> UV signature
mutations.
7.
➢Prognosis:
• No cure,genetic counselling
• Sun avoidance, broad spectrum sunscreens (preferably tinted)
• Physical methods of sun protection
• Vit D supplements
• Cancer screening every 3 months
• Existing lesions (AK, SK,Keratoacanthomas)-treat with cryo/5
FU/imiquimod.
• Isotretinoin to prevent development of cancers.
• Ophthal and neuro referral.
8.
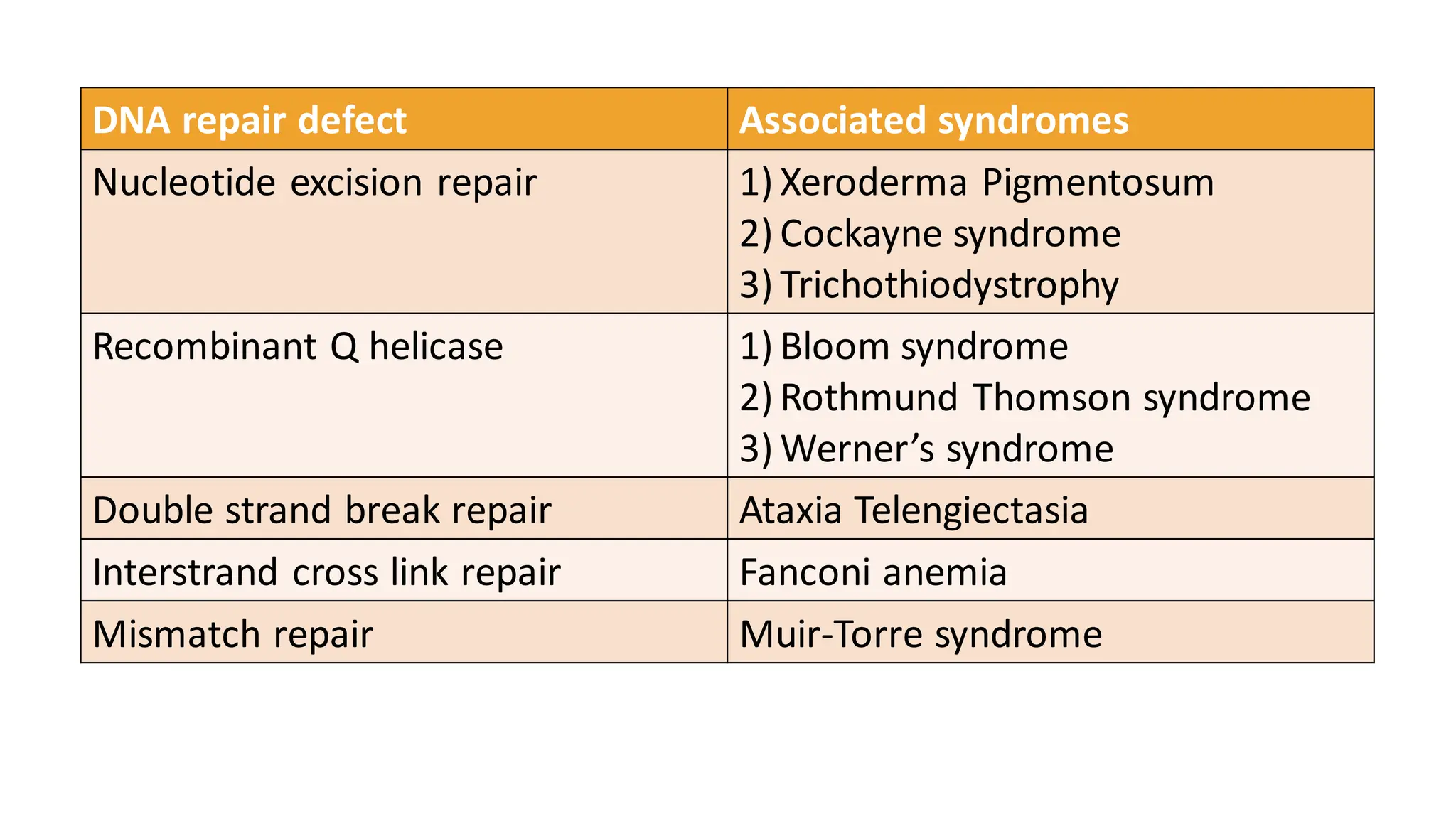
• Newer therapies-topical liposomal preparation of microbial DNA
repair enzyme T4 endonuclease, Sulphonylureas (glimiperide,
acetohexamide), Nicotinamide
9.
Cockayne syndrome
• NeillDingwall syndrome
• AR
• Defective TC-NER (RNA synthesis does not resume after UVR)
• Genes involved- CSA (ERCC8), CSB (ERCC6)
• CSA (20%) is milder form than CSB (80%)
• 3 clinical subtypes:
- CS1- classic
- CS2- severe
- CS3- late onset, milder
10.
➢ Clinical features:
-Photosensitivity (present since birth,improves)
- short stature, sparse, often gray hairs
- typical mickey mouse like facies
(beaked nose, large ears,
enophthalmia,prognathism)
- cachexia (cachexic dwarfism)
- SNHL/CHL
- Ophthal- salt and pepper retina, cataracts, optic atrophy
- CNS- progressive cognitive decline, extensive
demyelination, spasticity, choreoathetosis (basal
ganglia calcification).
11.
• No increasedrisk of skin cancers.
• Dental caries (86%)
• Patients die by 4th decade due to neurological complications.
• XP/CS overlap- XPB, D, F, G genes are involved.
• Variants- COFS, UV sensitivity syndrome
➢ Diagnosis:
• CT/MRI brain- loss of white matter and ventricular dilatation are
the earliest findings, tigroid leukodystrophy
• Prenatal diagnosis by amniocentesis and CVS to look for defective
gene
• Postnatal- UV irradiated fibroblasts obtained from skin biopsy are
cultured for 24 hrs, no RNA synthesis and normal UDS provides
diagnosis.
12.
TRICHOTHIODYSTROPHY
• Tay syndrome
•AR
• Defect in DNA repair (NER) as well as transcription.
• 2 types :
1) TTD-P (photosensitive):genes involved- ERCC2 (XPD),
ERCC3 (XPB) and GTF2H5 (TTDA), involved in transcription
repair protein IIH complex.
2) TTD-N (non-photosensitive):10-20%, gene involved-
MPLKIP
13.
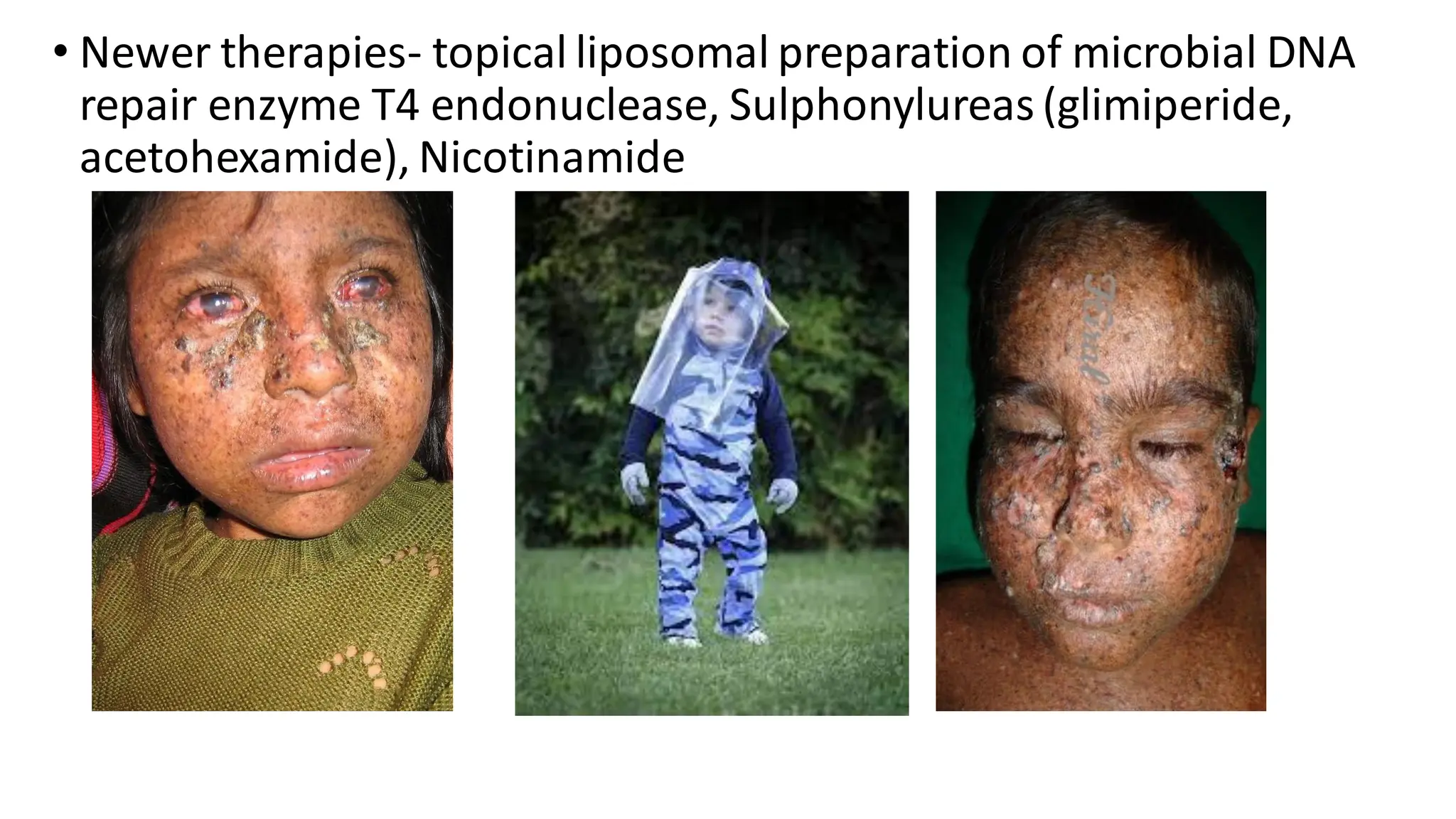
➢ Clinical features:
•Photosensitivity, no risk of skin ca.
• Icthyosis (TTD-P), often resembles CIE.
• Hair – short, sparse hairs with ‘tiger tail abnormality’ in hair
shafts under polarized microscopy (due to decrease sulphates
in hair), alopecias
• Nails- dystrophic
• CNS- intellectual disability, ataxia
• Hypogonadism, decreased fertility
• Others- abnormal facies (micrognathia, large ears), PPK, KP,
cataracts, osteosclerosis,hypogammaglobulinemia with
recurrent infections.
14.
• XP/TTD overlap-ERCC2 (XPD), ERCC3 (XPB) involvement
• D/D- Cockayne syndrome, Menkes Kinky hair disease, XP, Netherton
syndrome.
• Death is early due to severe infections.
15.
Rothmund- Thompson Syndrome
•Poikiloderma congenitale
• AR
• Gene involved- RECQL4 (recombinant DNA helicase)
• DNA repair + replication are defective
➢ Clinical features:
• Photosensitivity (earliest manifestation), presents as
persistent erythema and edema of V area of neck, face and
extremities.
• Erythema resolves> mottled pigmentation, atrophy and
telangiectasia (poikiloderma)develops, also over covered
parts.
16.
• Growth retardationin seen.
• Abnormal bird like facies with sparse or absent hair over scalp,
eyelashes, eyebrows.
• Intelligence is normal
• Ophthal- Bilateral, early onset (within 3-7 years), anterior
subcapsularcataracts, bilateral glaucoma, keratoconus,retinal
coloboma
• Skeletal- thumb hypoplasia/absence,absent radii, brachydactyly,
clinodactyly, syndactyly, osteoporosis
17.
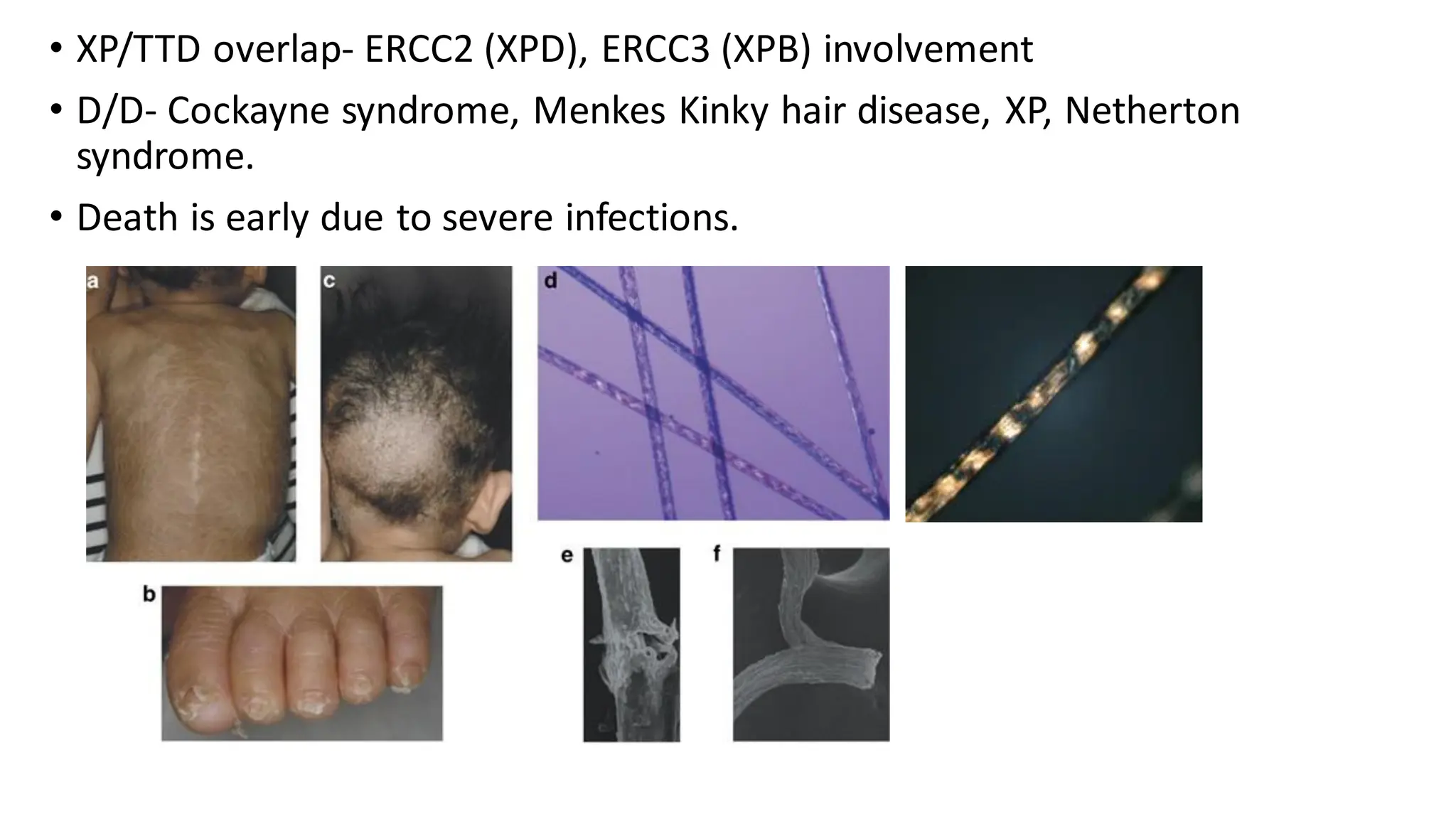
• Malignancies- M/Cis multicentric osteosarcomas,others
include SCC, BCC, Bowen’s disease, AML, Hodgkin’s lymphoma
• D/D- DKC, Werner, Kindler, Bloom, XP, Cockayne
• Treatment- Photoprotection, removal of hyperkeratotic
lesions (cryo, 5FU)
Vascular pulsed laser for poikiloderma, regular follow up for
cataracts cutaneous and systemic malignancies.
18.
Bloom Syndrome
• Congenitaltelangiectatic erythema with stunted growth
• AR
• Gene involved- RECQL3 (DNA helicase)
• High rate of sister chromatid exchange
• Common in Ashkenazi Jews
➢Clinical features:
• Photosensitivity over butterfly area of face
• Marked erythema and telengiectasia
• CALM
• Markedly stunted growth, may present as IUGR
19.
• Abnormal facies-dolicocephaly, keel shaped face, low set ears,
sharp nose
• Shrill voice
• Intelligence- subnormal
• Immunodeficiency ( IgM,IgA,IgG) associated recurrent,
chronic diarrhea, respiratory infections (often fatal)
• Malignancies- cutaneous as well as systemic (lymphoreticular
are common, visceral malignancies, SCC, BCC, Bowen’s
disease)
• Ocular- conjunctivitis, telengiectasias
20.
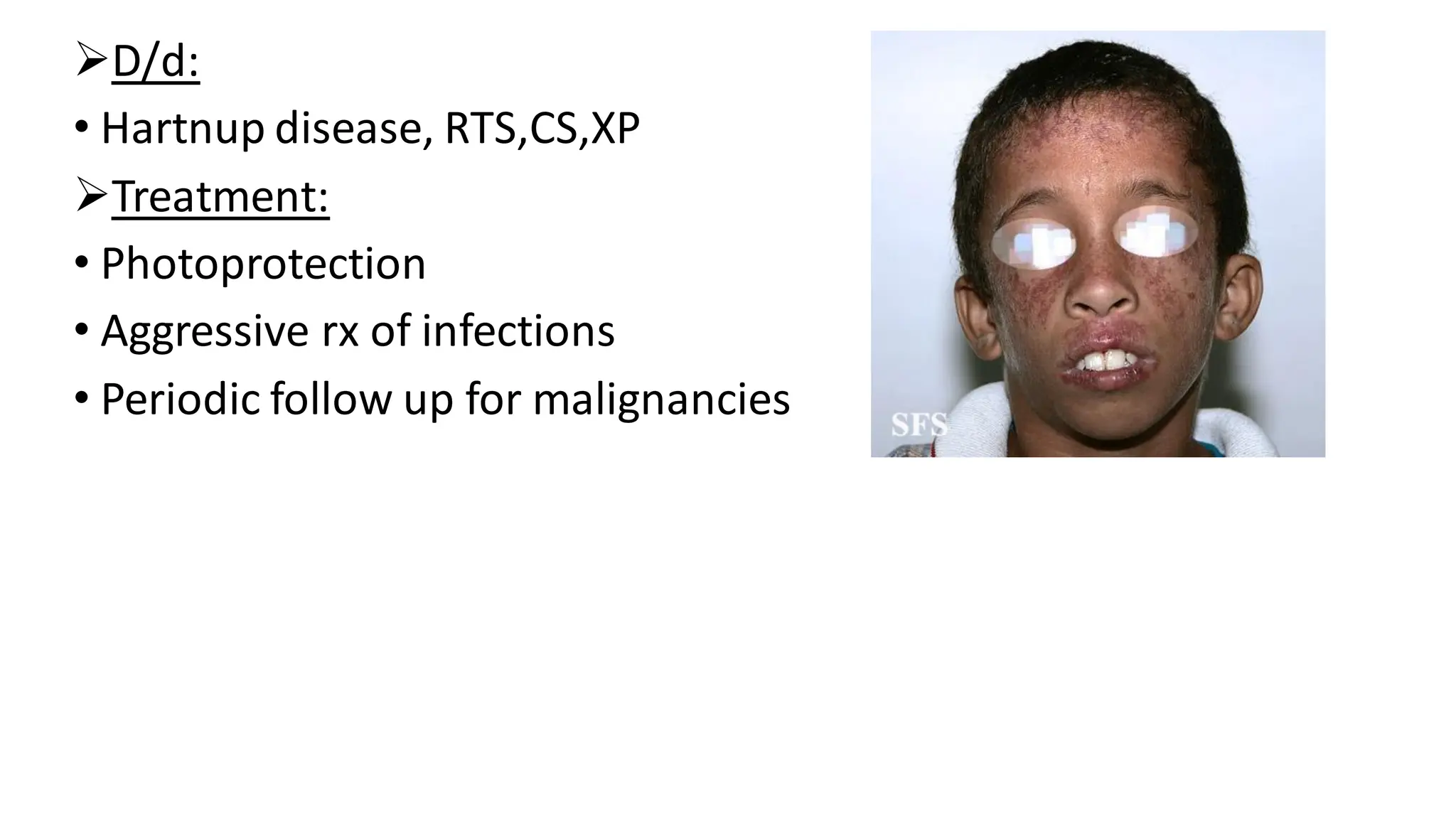
➢D/d:
• Hartnup disease,RTS,CS,XP
➢Treatment:
• Photoprotection
• Aggressive rx of infections
• Periodic follow up for malignancies
21.
Ataxia Telangiectasia
• AR
•Gene involved- ATM (Ataxia telengiectasia mutated) gene
➢ Clinical features:
• Ataxia and eye movement abnormalities appear in toddler or
school going age
• Movement abnormalities- inability to sit or stand straight,
swaying, ataxic gait
• Defective hand eye co-ordination- difficulty in writing, eating
• Nystagmus is primary and lateral gaze.
• Neurological abnormalities are non progressive beyond 12-
15 years of age
22.
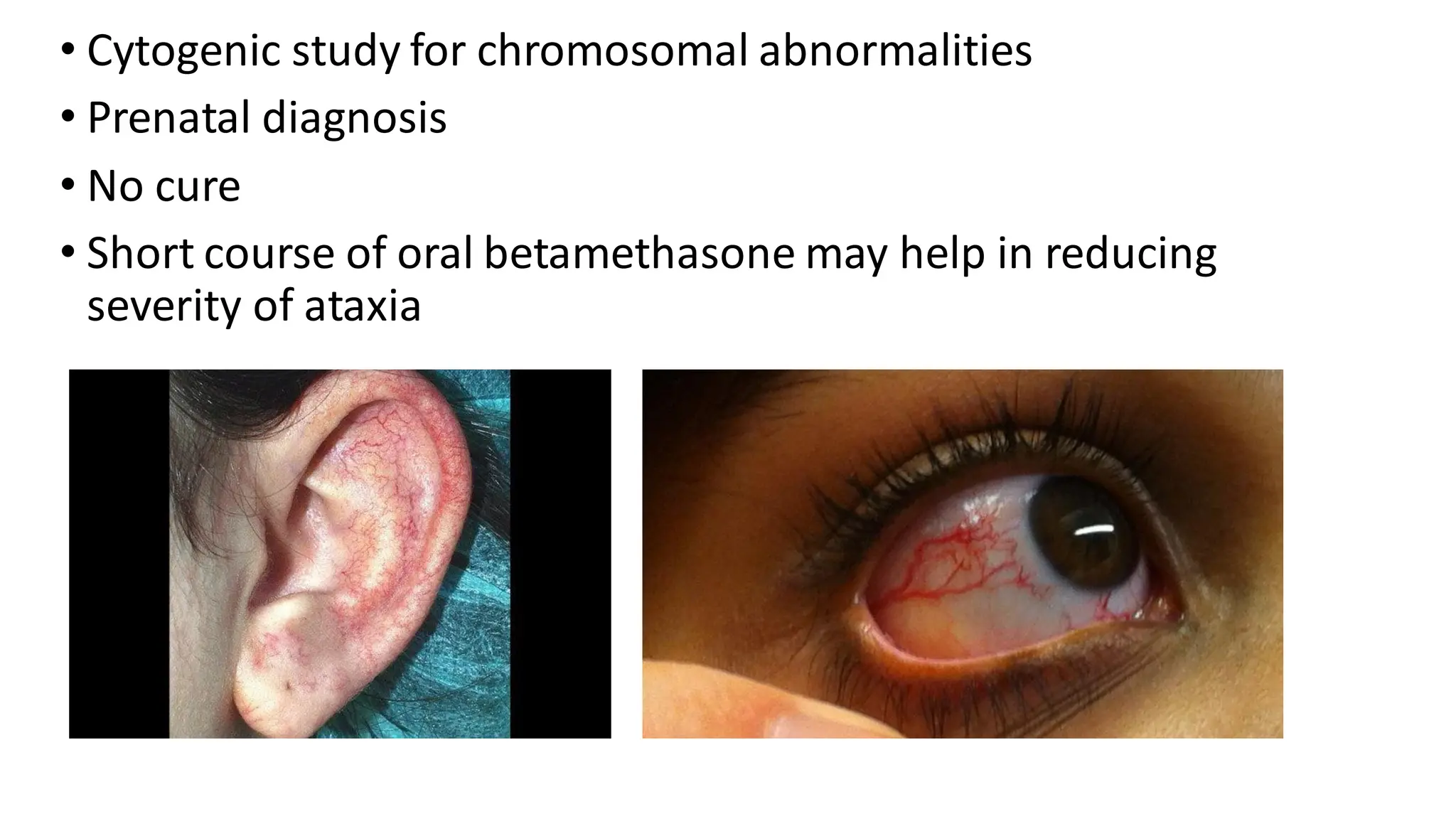
• Dystonia, choreoathetosis,myoclonic jerks may be present
• Telengiectasias appear at 5-8 years of age
• Oculo-muco cutaneous, most obvious in bulbar conjunctiva
• Accentuation on sun exposure
• Immunodeficiency- recurrent sino-pulmonaryinfections
➢ Investigations:
• Raised CEA, alpha-fetoprotein in all patients (may help in
diagnosis after 2 years of age)
• Decreased IgM, IgA, IgG
• MRI brain and spinal cord- cerebellar degeneration
23.
• Cytogenic studyfor chromosomal abnormalities
• Prenatal diagnosis
• No cure
• Short course of oral betamethasone may help in reducing
severity of ataxia
24.
Fanconi’s anemia
• AR
•Increased cellular sensitivity to interstrand cross link agents
like mitomycin C.
• Early onset bone marrow failure with increased
predispositionto systemic malignancies (leukemia,
lymphoma, oesophageal ca, SCC of head and neck)
• 2/3rd of patients have congenital malformations of kidneys,
heart, skeletal system (absent thum/radii).
• Cutaneous- CALM, reticulate or patchy pigmentation
• Abnormal facies- small head, mouth
25.
Muir-Torre Syndrome
• AD
•Defect in DNA mismatch repair (MSH2 gene)
• Phenotypic variant of hereditary non polyposis colorectal cancer (Lynch
syndrome)
• Multiple sebaceous neoplasms (sebaceous ademas/carcinomas,
epitheliomas), keratoacanthomas,BCC
• Visceral malignancies- m/c is colorectal ca
• Colonoscopy to be done every 1-2 years starting from 25 years of 5 years
before the earliest age of diagnosis of colorectal ca in the family, annually
after 40 years.
• Prophylactic colectomy in pts with confirmed gene mutation.
• Sebaceous neoplasms below the neck- suspect Muir-Torre syndrome.
27.
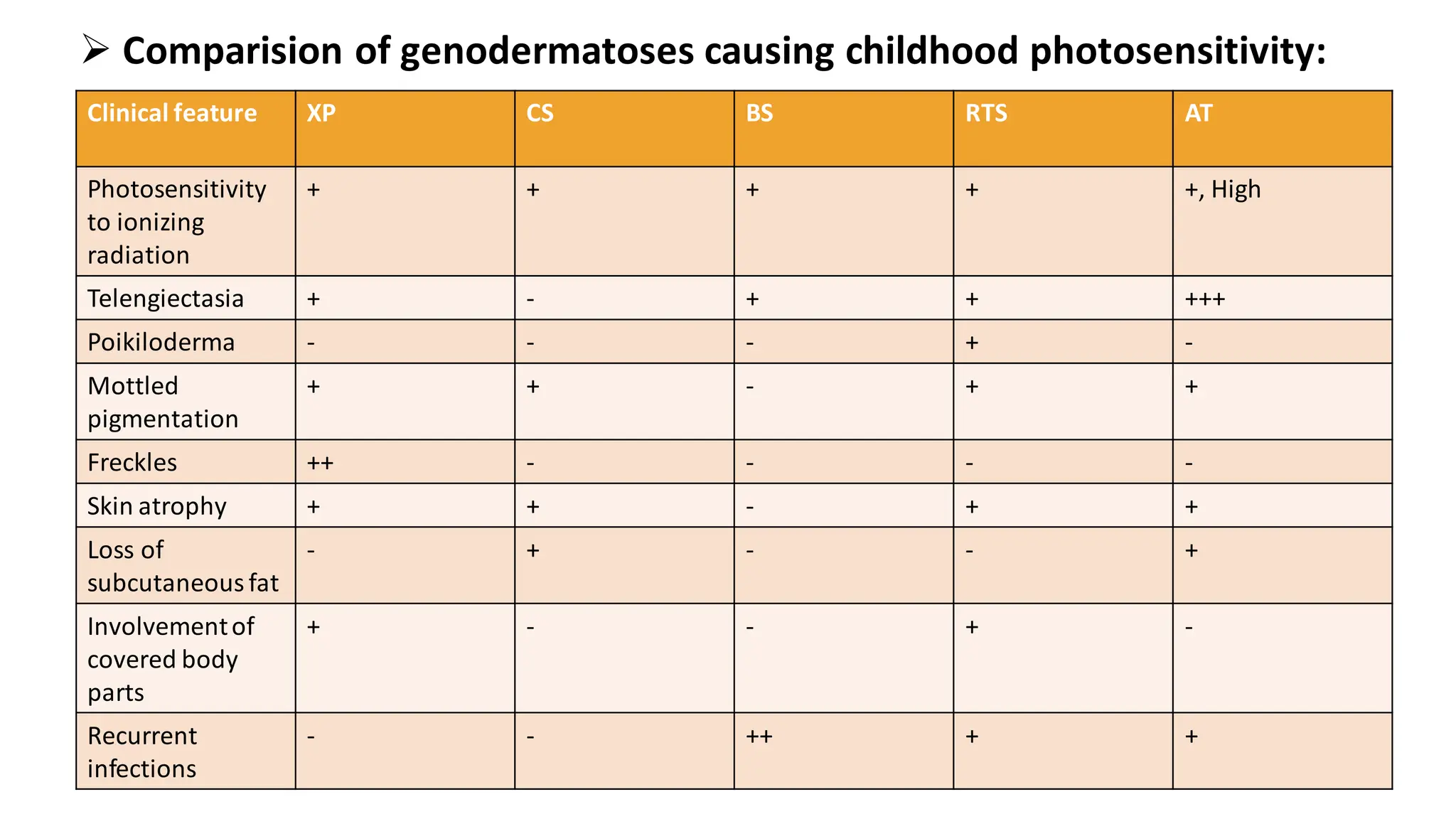
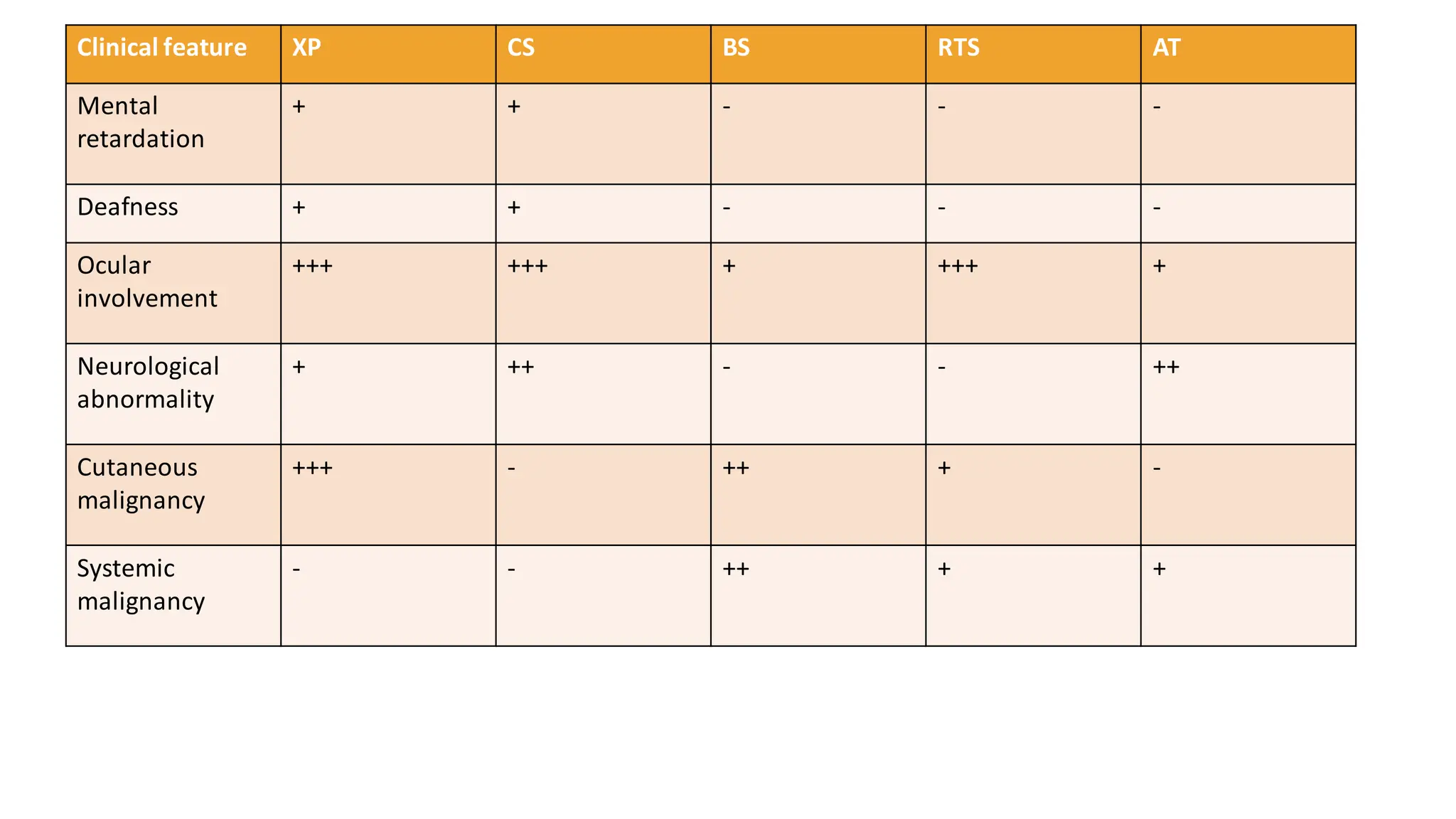
➢ Comparision ofgenodermatoses causing childhood photosensitivity:
Clinical feature XP CS BS RTS AT
Photosensitivity
to ionizing
radiation
+ + + + +, High
Telengiectasia + - + + +++
Poikiloderma - - - + -
Mottled
pigmentation
+ + - + +
Freckles ++ - - - -
Skin atrophy + + - + +
Loss of
subcutaneousfat
- + - - +
Involvementof
covered body
parts
+ - - + -
Recurrent
infections
- - ++ + +