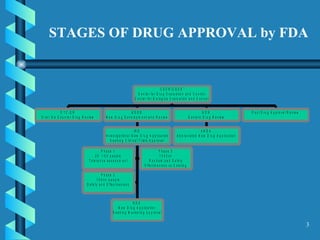
The document outlines the key stages and requirements for drug development and approval by the FDA. It discusses the necessary chemical, manufacturing and controls (CMC) information that must be submitted at each stage, including details on the drug substance, manufacturing process, specifications, stability testing and other information for both the drug substance and drug product.