Download to read offline


















![Morphology of Infiltrating
Astrocytomas
• Histologic differentiation (WHO grades II to IV)
correlates well with the clinical course:
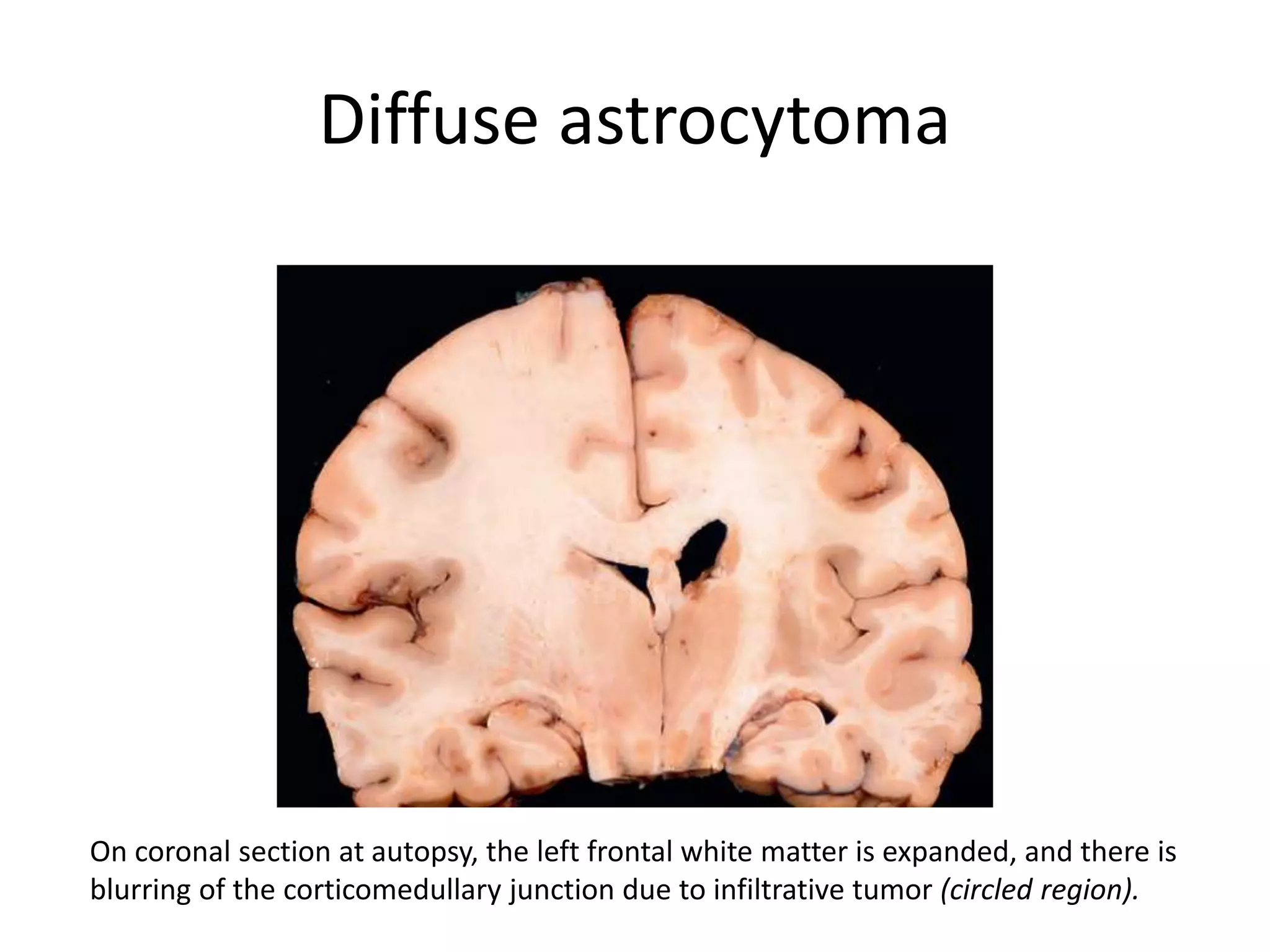
– Diffuse astrocytomas (grade II)
– Anaplastic astrocytomas (grade III)
– Glioblastomas (grade IV; previously called
glioblastoma multiforme [GBM])](https://image.slidesharecdn.com/centralnervoussystem3-210811141651/75/Central-nervous-system-Tumors-20-2048.jpg)



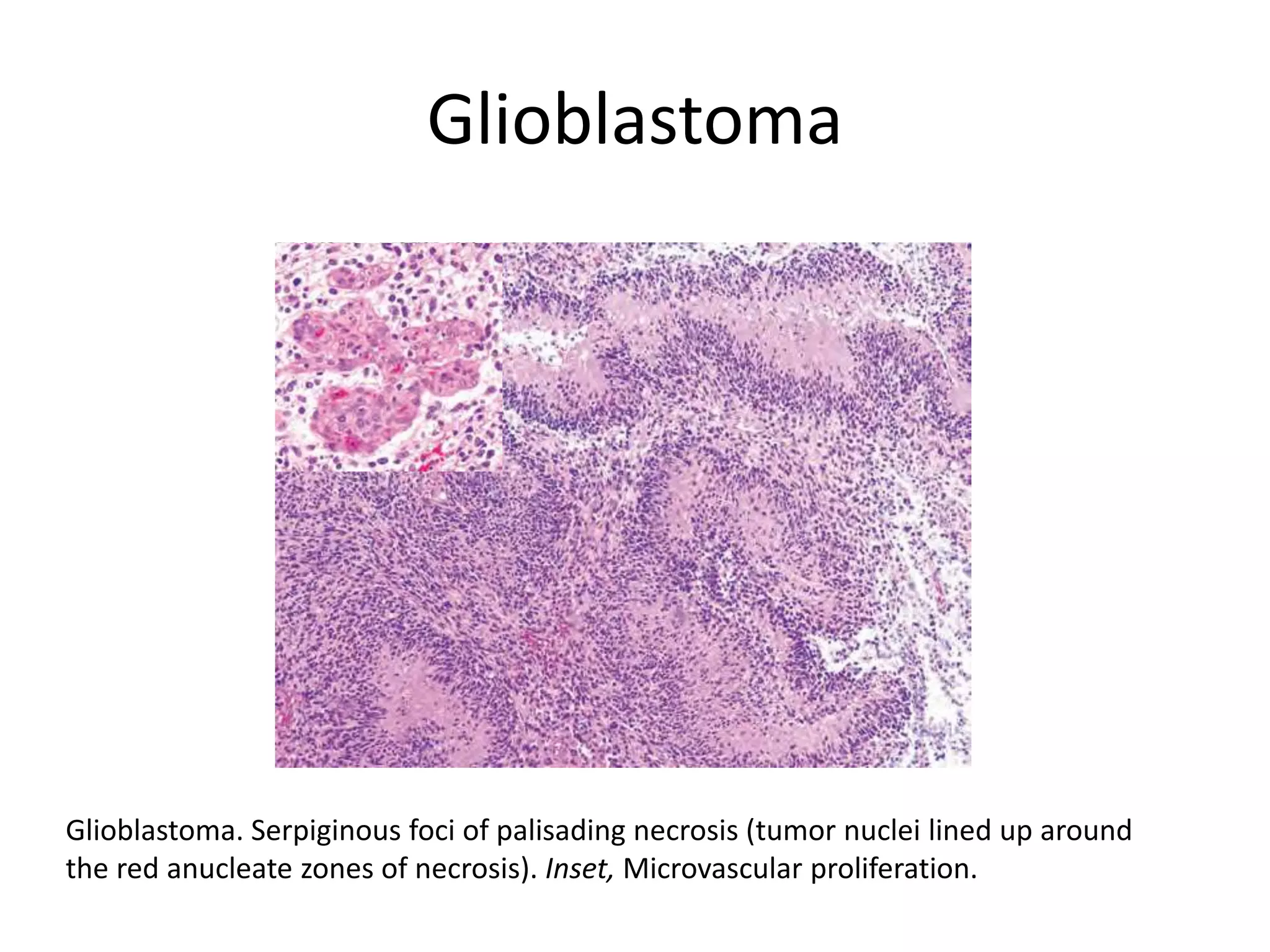
![Glioblastomas (grade IV; previously called
glioblastoma multiforme [GBM])
• Composed of a mixture of firm white areas,
softer yellow foci of necrosis, cystic change,
and hemorrhage;
• There is also increased vascularity.
• Increased tumor cell density along the
necrotic edges is termed pseudopalisading.](https://image.slidesharecdn.com/centralnervoussystem3-210811141651/75/Central-nervous-system-Tumors-25-2048.jpg)












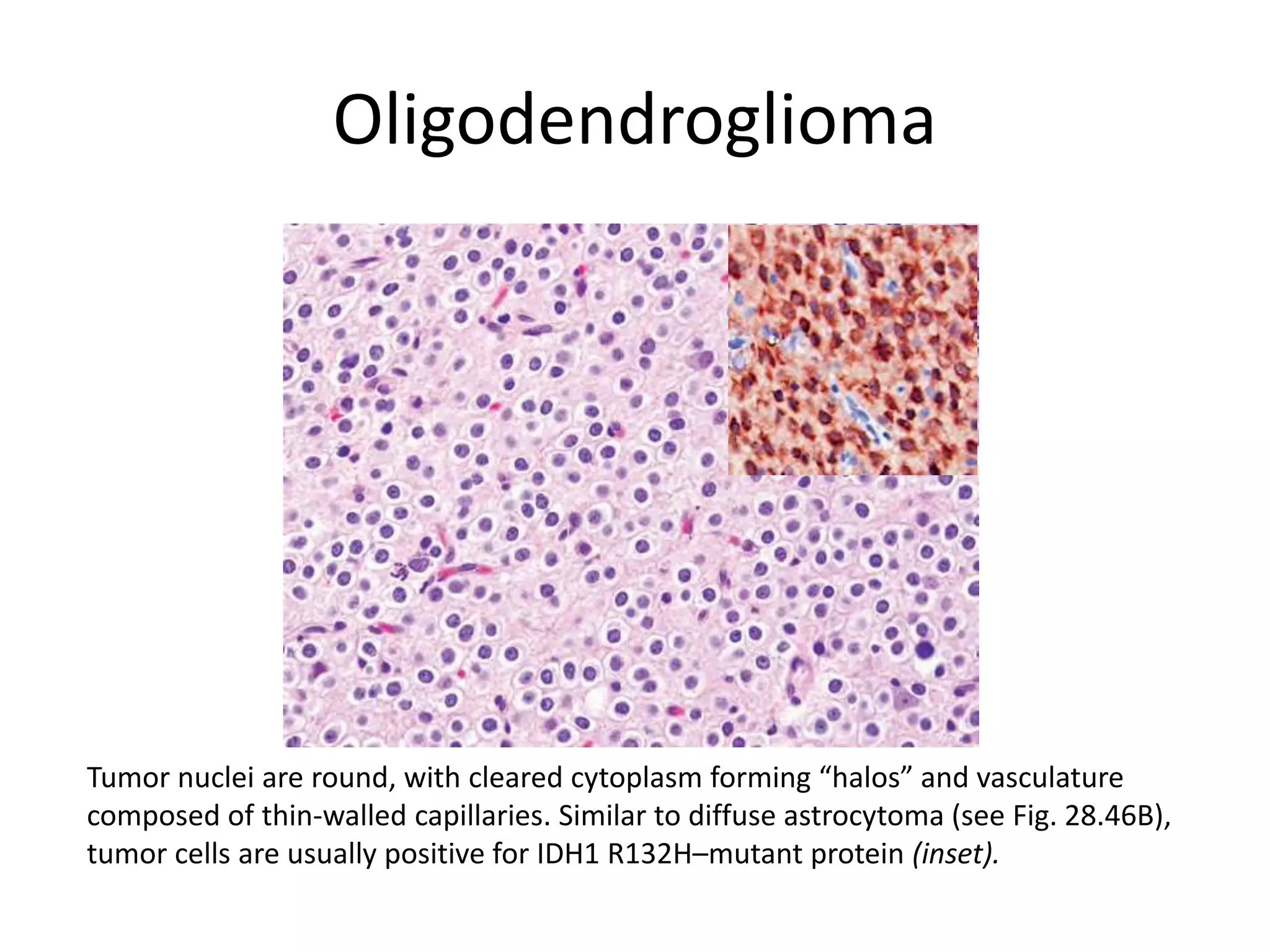



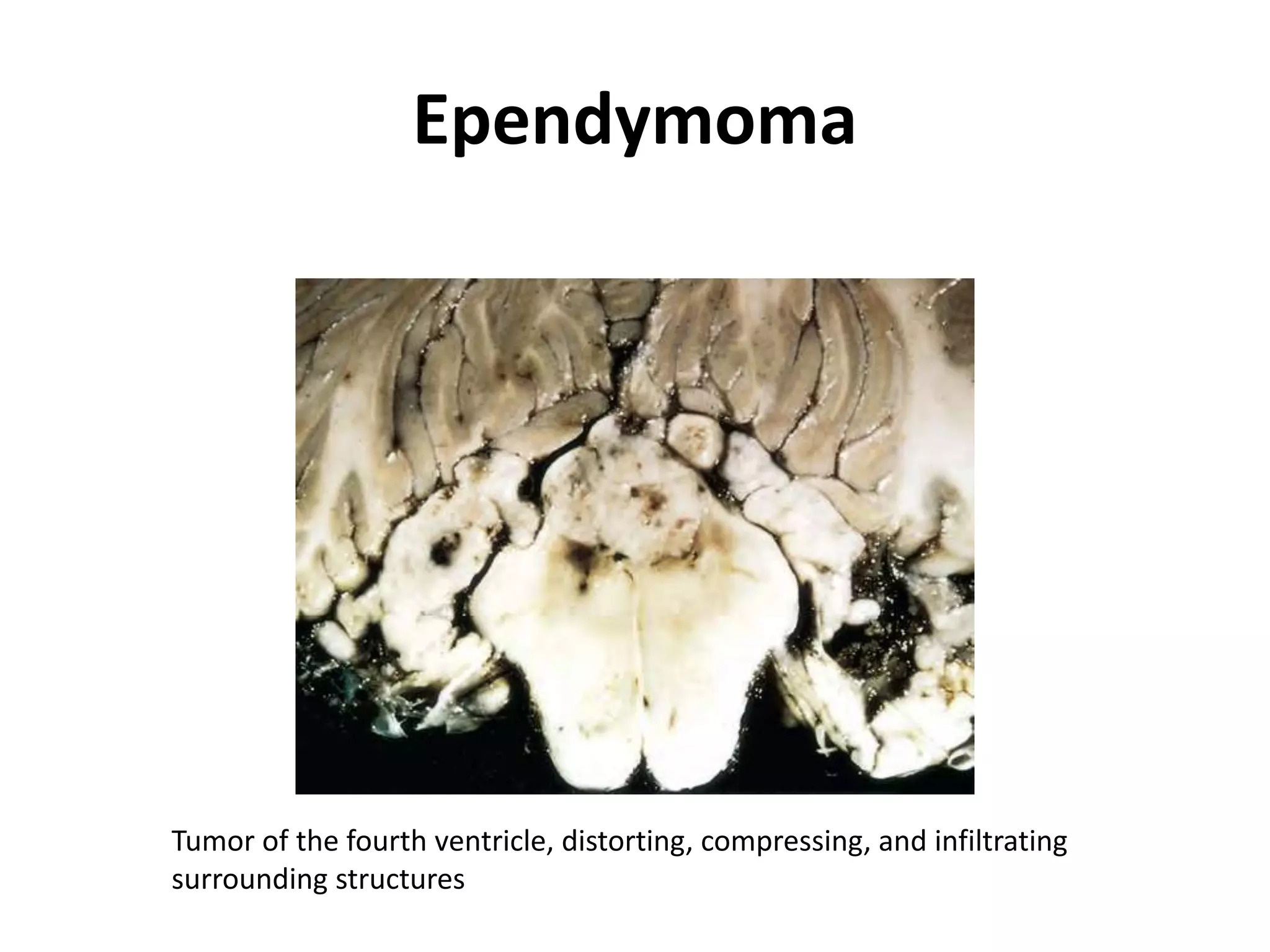








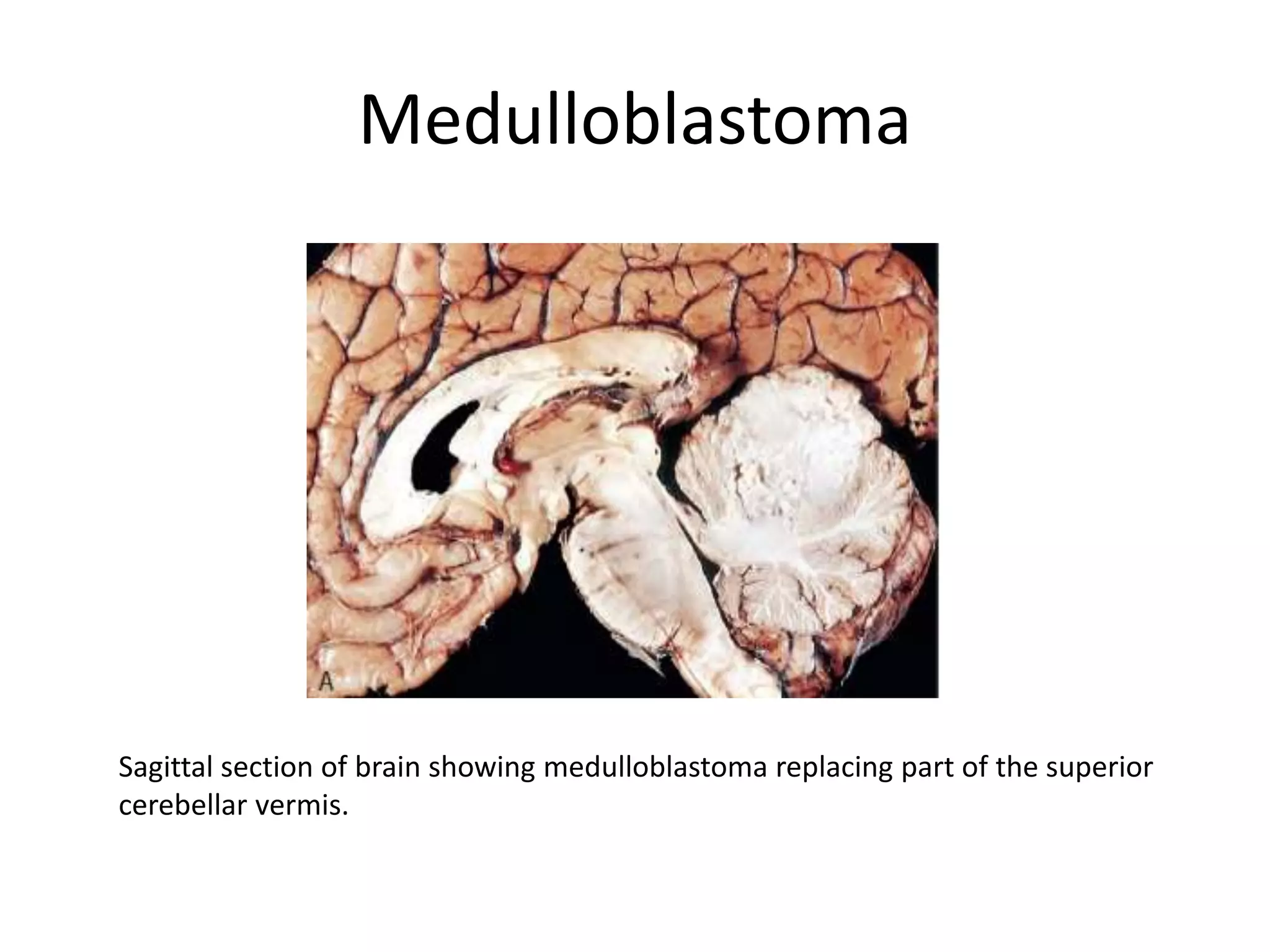



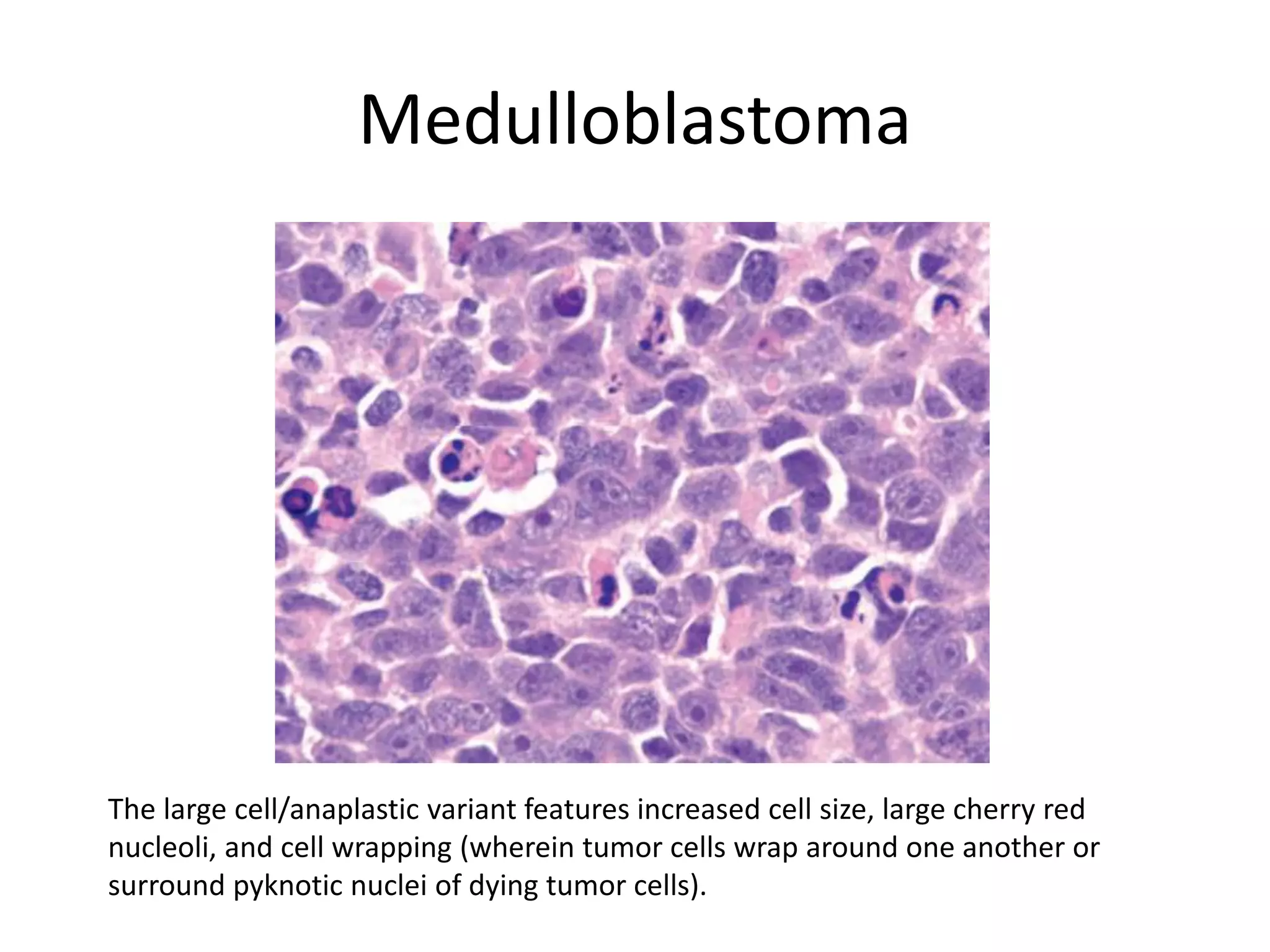











This document discusses various tumors of the central nervous system (CNS). It provides details on: - The classification and incidence of different CNS tumors. Primary CNS tumors account for 20% of childhood cancers, with 70% occurring in the posterior fossa. - The molecular genetics and morphology of common tumor types, including gliomas, astrocytomas, oligodendrogliomas, ependymomas, and medulloblastomas. Factors like location, growth patterns, and genetic mutations influence tumor behavior. - The clinical features and prognosis of different tumor types. More malignant tumors are associated with worse outcomes, though treatments like surgery and radiation can improve survival for some tumors. Location within the



































































