THE CELL ANDIT’S FUNCTION
100 TRILLION CELLS IN THE HUMAN BODY
CELLLS ARE BUILDING BLOCKS OF THE BODY PROVIDING STRUCTURE
for the body’s tissues and organs, ingesting nutrients and
converting them to energy, and performing specialized
functions.
CELL- BASIC UNIT OF LIFE.
3.
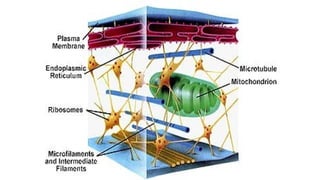
CELL ORGANIZATION
• TYPICALCELL CAN BE SEEN THROUGH LIGHT MICROSCOPE
• TWO MAJOR PARTS: NUCLEUS AND CYTOPLASM
• DIFFERENT SUBSTANCES THAT MAKE UP THE CELL ARE COLLECTIVELY KNOWN AS
PROTOPLASM
• PROTOPLASM INCLUDES: WATER( 70-85%), IONS, PROTEINS( STRUCTURAL AND FUNCTIONAL),
LIPIDS AND CARBOHYDRATES.
• INTRACELLULAR AND EXTRACELLULAR COMPARTMENTS.
• INTRACELLULAR ORGANELLES- HIGHLY ORGANISED PHYSICAL STRUCTURES INSIDE THE CELL.
4.
Continued….
• THE CELLRECEIVES MESSAGES OR INSTRUCTIONS ON PROTEIN SYNTHESIS
THROUGH FIRST MESSENGERS/LIGAND
• THE LIGAND (HORMONE OR NEUROTRANSMITTER) BINDS TO A RECEPTOR
ON THE CELL MEMBRANE
• THE MESSAGE IS THEN CARRIED INTO THE CELL THROUGH A SIGNAL
TRANSDUCTION MECHANISM INVOLVING SECOND MESSENGERS
• ONCE THE MESSAGE REACHES THE NUCLEUS, THE CELL WILL RESPOND BY
SYNTHESIZING A PROTEIN
• THE PROCESS OF PROTEIN SYNTHESIS IS CARRIED OUT SYSTEMATICALLY
FROM TRANSCRIPTION TO RELEASE OF A FUNCTIONAL PROTEIN BY THE CELL
5.
CONTD..
• A FAILUREIN THE RECEPTIN AND TRANSDUCTION OF THESE INSTRUCTIONS
AT ANY LEVEL MAY INTERFERE WITH THE PROCESS OF PROTEIN SYNTHESIS
• THE PROTEIN MAY NOT BE FORMED AT ALL; IT MAY BE FORMED BUT IS OF
AN ABNORMAL STRUCTURE; MAY BE FORMED BUT IN INADEQUATE
QUANTITIES
• THIS WILL RESULT IN DIFFERNER OF ABNORMAL OR ABSENTT PROTEINS
SUCH AS ENZYME/HORMONE DEFICINCIES; ABNORMAL RECEPTORS ETC.
• AN IMPAIRMENT INVOLVING ANY OF THE ORGANELLES INVOLVED IN THE
PROCESS OF PROTEIN SYNTHESIS MAY RESULT IN SIMILAR PATHOLOGICAL
CONDITIONS
7.
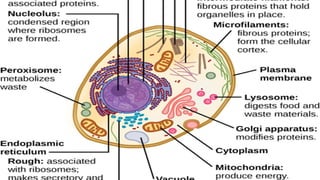
CELL MEMBRANE( PLASMAMEMBRANE)
• ENVELOPES THE CELL.
• APPROXIMATE COMPOSITION: PROTEINS 55%, PHOSPHOLIPIDS 25%, CHOLESTEROL
13%, OTHER LIPIDS 4%, CARBORHYDRATES 3%
• BASIC STRUCTURE IS A LIPID BILAYER( THIN, DOUBLE LAYERED FILM EACH LAYER ONE
MOLECULE THICK THAT IS CONTINOUS OVER THE ENTIRE CELL SURFACE AND
INTERSPERSED IN IT ARE LARGE GLOBULAR PROTEINS.
• LIPID BILAYER: 3 MAIN LIPIDS( PHOSPHOLIPIDS- MOST ABUNDANT, SPHINGOLIPIDS
AND CHOLESTEROL)
8.
CELL MEMBRANE
• PHOSPHOLIPIDMOLECULE- PHOSPHATE END-HYDROPHILIC, FATTY END-
HYDROPHOBIC. HYDROPHOBIC ENDS REPELLED BY WATER BUT ARE MUTUALLY
ATTRACTED TO ONE ANOTHER THUS ATTRACTING EACH OTHER IN THE MIDDLE OF
THE MEMBRANE.
• LIPID BILAYER IS IMPERMIABLE TO USUAL WATER SOLUBLE SUBSTANCES E.g. ions,
glucose and urea. Conversely fat soluble substances e.g. oxygen, carbon dioxide and
alcohol can penetrate with ease.
9.
Cell membrane
• Cellmembrane proteins: integral that protrude all the way through the membrane,
peripheral- attached only to one surface of the membrane and do not penetrate.
• Integral- provide structural channels(pores) through which water and water soluble
molecules can diffuse between extracellular and intracellular fluids. Act also as carrier
proteins for transporting substances that cannot penetrate lipid bilayer. Serve as
receptors for water soluble hormones such as peptide hormones.
• Peripheral- attached to integral proteins. function almost entirely as enzymes or as
controllers of transport of substances.
10.
Cell membrane
• Membranecarbohydrates- occur almost invariably in combination with proteins or
lipids in form of glycolipids or glycoproteins.
• Proteoglycans- carbohydrate compounds bound to small protein cores and are loosely
attached to outer surface of the well thus the entire outside surface of the cell often
has a loose carbohydrate coat called glycocalyx.
11.
Cell membrane
• Thecarbohydrate moieties attached to the outer surface of the cell have several
important functions:
1. Many of them have a negative electrical charge, which gives most cells an overall
negative surface charge that repels other negatively charged objects.
2. The glycocalyx of some cells attaches to the glycocalyx of other cells, thus attaching cells
to one another.
12.
Cell membarne
• 3.Many of the carbohydrates act as receptor substances for binding hormones, such
as insulin; when bound, this combination activates attached internal proteins that, in
turn, activate a cascade of intracellular enzymes.
• 4. Some carbohydrate moieties enter into immune reactions,
13.
Endoplasmic reticulum
• InterconnectedTubular and flat vesicular structures.
• Processes molecules made by the cell and transports them to their
specific destinations inside or outside the cell.
Two types: granular and agranular
• Granular e.r- ribosomes attached to the e.r. the ribosomes are
composed of a mixture of r.n.a and proteins and function to
synthesize new protein molecules in the cell
• Agranular e.r.- functions for the synthesis of lipid substances mainly.
16.
Golgi apparatus
• Closelyrelated to e.r. has membranes similar to those of Agranular e.r.
• Composed of a stuck of 4 or more layers of thin, flat enclosed vesicles lying near one
side of the nucleus. Prominent in secretory cell.
• Functions in association with e.r
• “transport vesicles” (also called endoplasmic reticulum vesicles, or ER vesicles)
continually pinch of from the endoplasmic reticulum and shortly thereafter fuse with
the Golgi apparatus. In this way, substances entrapped in the ER vesicles are
transported from the endoplasmic reticulum to the Golgi apparatus. The transported
substances are then processed in the Golgi apparatus to form lysosomes, secretory
vesicles, and other cytoplasmic components
17.
lysosomes
• Vesicular organellesthat form by breaking off from golgi apparatus and then
dispersing throughout cytoplasm. Has a typical lipid bilayer membrane, and filled with
granules containing digestive(hydrolase) enzymes as many as 40.
• Intracellular digestive system. digest (1) damaged cellular structures, (2) food particles
that have been ingested by the cell, and (3) unwanted matter such as bacteria.
18.
peroxisomes
• Physically similarto lysosomes but different in two ways
• Believed to be formed by self replication or perhaps by budding off fro the smooth e.r. rather
than from golgi
• They contain oxidases rather than hydrolases. E.g hydrogen peroxide, catalase(For instance,
about half the alcohol a person drinks is detoxified into acetaldehyde by the peroxisomes of
the liver cells in this manner)
mitochondria
• Powerhouse ofthe cell. Without them all cellular functions of the
cell would cease.
• Their numbers depend on the cell function.
• Composed of inner and outer membrane. Inner membrane folded to
form cristae( increase surface area) onto which oxidative enzymes
are attached. Inner cavity filled with a matrix that contains large
quantities of dissolved enzymes necessary for extracting energy from
nutrients.
• Main function- production of atp. They are self replicative. Has its
own dna.
21.
Cell cytoskeleton- filamentand tubular
structures
• Cell cytoskeleton- network of fibrillary proteins organized into
filaments or tubules.
• The cytoskeleton is made up primarily of microtubules, intermediate
filaments, and microfilaments
• proteins and organelles move along microtubules and
microfilaments from one part of the cell to another, propelled by
molecular motors.
22.
Cell cytoskeleton
• Microtubules-madeup of two globular protein subunits: α- and β-
tubulin. A third subunit, y-tubulin, is associated with the production
of microtubules by the centrosomes.
• They provide the tracks along which several different molecular
motors move transport vesicles, organelles such as secretory
granules, and mitochondria from one part of the cell to another.
They also form the spindle, which moves the chromosomes in
mitosis. Cargo can be transported in either direction on
microtubules.
• There are several drugs that disrupt their function e.g. colchicine,
paclitaxel, vinblastine.
23.
Cell cytoskeleton.
• Intermediatefilaments- made up of various sub-units.
• form a flexible scaffolding for the cell and help it resist external
pressure.
• In their absence, cells rupture more easily, and when they are
abnormal in humans, blistering of the skin is common. proteins that
make up intermediate filaments are cell-type specific, and are thus
frequently used as cellular markers. E.g. vimentin is a major
intermediate filament in fibroblasts, whereas cytokeratin is
expressed in epithelial cells
24.
Cell cytoskeleton
• Microfilaments-made up of actin(It is the most abundant protein in
mammalian cells)
• Filamentous (F) actin refers to intact microfilaments and globular (G)
actin refers to the unpolymerized protein actin subunits.
• The actin filaments interact with integrin receptors and form focal
adhesion complexes, which serve as points of traction with the
surface over which the cell pulls itself. In addition, some molecular
motors use microfilaments as tracks.
25.
Molecular motors
• Thereare three super families of molecular motors: kinesin, dynein, and
myosin.
• Kinesin- double headed molecule that tends to move its cargo toward the “+”
ends of microtubules, and sometimes to the negative end of microtubules.
• Some are involved in meiosis and mitosis
• Dyneins- have two heads, with their neck pieces embedded in a complex of
proteins. have a function like that of conventional kinesin, except they tend to
move particles and membranes to the “–” end of the microtubules.
• Myosin- divided into 18 classes. The heads of myosin molecules bind to actin
and produce motion by bending their neck regions (myosin II) or walking along
microfilaments, one head after the other
26.
centrosomes
• Located nearthe nucleus. made up of two centrioles and surrounding
amorphous peri-centriolar material. The centrioles are short cylinders
arranged so that they are at right angles to each other.
• The centrosomes are microtubule-organizing centers (MTOCs) that contain
γ-tubulin. The microtubules grow out of this γ-tubulin in the pericentriolar
material. When a cell divides, the centrosomes duplicate themselves, and
the pairs move apart to the poles of the mitotic spindle, where they
monitor the steps in cell division. In multinucleate cells, a centrosome is
near each nucleus.
27.
cilia
• specialized cellularprojections that are used by unicellular organisms
to propel themselves through liquid and by multicellular organisms
to propel mucus and other substances over the surface of various
epithelia
28.
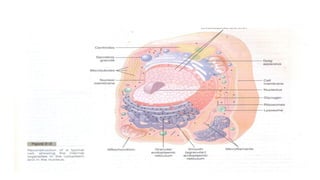
nucleus
• The nucleusis made up in large part of the chromosomes(occur in
pairs), the structures in the nucleus that carry a complete blueprint
for all the heritable species and individual characteristics of the
animal.
• Each chromosome made up of d.n.a( about 2 meters long) but it can
fit in the nucleus because at intervals it is wrapped around a core of
histone proteins to form a nucleosome( about 25 million in each
nucleus). The whole complex of DNA and proteins is called
chromatin.
• The ultimate units of heredity are the genes on the chromosomes.
29.
nucleus
• The nucleusof most cells contains a nucleolus ( a patchwork of
granules rich in RNA). Can be one or more.
• They are the site of synthesis of ribosomes( protein synthesis).
• The interior of the nucleus has a skeleton of fine filaments that are
attached to the nuclear membrane, or envelope, which surrounds
the nucleus. This membrane is a double membrane, and spaces
between the two folds are called perinuclear cisterns.
30.
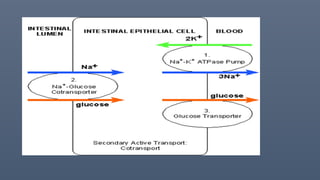
Transport across cellmembrane
• Primary pathways include exocytosis, endocytosis, movement through ion
channels, and primary and secondary active transport
• Exocytosis- Vesicles containing material for export are targeted to the cell
membrane where they bound via the v-SNARE/t-SNARE arrangement. The area
of fusion then breaks down, leaving the contents of the vesicle outside and the
cell membrane intact. This is the Ca2+-dependent process of exocytosis
• secretion from the cell occurs via two pathways: non-constitutive(regulated
pathway- processing of prohormones to mature hormones before exocytosis)
and constitutive pathway( no processing or storage)
31.
endocytosis
• The reverseof exocytosis
• Is of various types depending on size of particles being transported,
as well as the regulatory requirements for the particular process:
• phagocytosis, pinocytosis, clathrin-mediated endocytosis,
caveolae-dependent uptake, and nonclathrin/noncaveolae
endocytosis.
33.
Membrane permeability andmembrane
transport protiens.
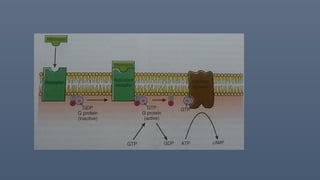
• Voltage gated,
• ligand gated- external or internal( ca2+, Cyclic amp, g-protein)
• When carrier proteins move substances in the direction of their
chemical or electrical gradients, no energy input is required and the
process is called facilitated diffusion.
• Active transport. na+-K+ atpase, H+-K= ATPASE.
• UNIPORTS, SYMPORTS, ANTIPORTS
Signal transduction
• Receptorsand ligands
• Intracellular calcium as a second messenger
• Ca2+-binding proteins have been described, including troponin,
calmodulin, and calbindin. Ca2+-binding proteins include: troponin,
calmodulin, and calbindin.
• G protein, IP3.
39.
Intercellular connections.
• Intercellularjunctions that form between the cells in tissues can be broadly split
into two groups: junctions that fasten the cells to one another and to
surrounding tissues, and junctions that permit transfer of ions and other
molecules from one cell to another
• Tight junction(zonula occludens), desmosomes and zonula adherens,
hemidesmosomes and focal adhesions.
• Gap junctions-two corresponding gap juctions line up with one another to allow
passage of substances between the cells without passing through e.c.f.
mutations in genes that code gap junctions can lead to disease. e.g.
predisposition to sudden cardiac death, female sterility, abnormal bone
development, abnormal growth in the liver, cataracts, hearing loss, and a host of
other abnormalities
DEFINITION
• Pathophysiology :Study of the underlying changes in body physiology
(molecular, cellular & organ system) occurring due to disease/injury
Cellular mechanisms pathophysiology encompasses:
• Cellular response to stress and noxious stimuli
• Cellular adaption to stress
• Cell injury and cellular death
• Causes of cell injury
• Effects of cell injury on cellular mechanisms
47.
Cellular response tostress and noxious stimuli
• Cells normally maintain a steady state called homeostasis; intracellular
milleu is kept within a fairly narrow range of physiologic parameters
• Physiologic stresses or stimuli, may lead to functional and structural changes
so as to achieve a new steady state (adaptation)
• Within certain limits, injury is reversible and cells return to a stable baseline
• If the adaptive capability is exceeded or if external stress is inherently
harmful, irreversible cell injury and cell death ensue
48.
Causes of cellinjury
1. Hypoxia (deficiency of oxygen) : Ischemia or anemia
2. Physical agent: burns, deep cold, radiation, electric shock and mechanical trauma
3. Biological agents: viruses, bacterial toxins, fungi and parasites
4. Chemical agents and drugs
5. Endogenous toxins :uremia, jaundice and diabetic
6. Immunologic reactions (hypersensitivity)
7. Nutritional imbalance :PEM, starvation, obesity, DM etc
8. Genetic abnormalities :Down syndrome & sickle anaemia
9. Ageing
49.
Mechanisms of cellinjury
• Depletion of ATP
• Mitochondrial damage
• Influx of calcium and loss of calcium homeostatis
• Accumulation of oxygen-derived free radicles (oxidative stress)
• Defects in membrane permeability
• Damage to DNA and proteins
50.
Cell injury: ATPDepletion
• May occur due to hypoxic or chemical (toxic) injury
• ATP production is in two ways:
• Oxidative phosphorylation of ADP in mitochondria
(main)
• Glycolytic pathway (anaerobic; uses glucose or
glycogen)
51.
ATP Depletition Cont...
•Consequences
1. Activity of plasma membrane energy dependent Na+, K+-ATPase) is
reduced; causes intracellar accumulation of Na+ and K+ diffusion out of the
cell
2. Cellular energy metabolism is altered – Cell switches to anaerobic glycolysis
(to maintain cell’s energy sources from ATP)
3. Failure of Calcium pumps – Influx of Ca2+ leading to damage of organelles
4. Structural disruption of protein synthetic apparatus; detachment of
ribosomes from rough ER and dissociation of polysomes
52.
ATP DePletition cont..
4.Misfolding of proteins and accumulation in the ER causing unfolded
protein response- may lead to cell injury and cell death
5. Irreversible damage to mitochondrial and lysosomal membranes
leading to necrosis
Mitochondrial Damage
• Consequences
1.Loss of mitochondrial membrane potential through formation of
mitochondrial permeability transion pore (leads to failure of
oxidative phosphorylation, depletion of ATP and cell necrosis)
2. Abnormal oxidative phosphorylation leading to formation of
reactive oxygen species, ROS
3. Sequestration of mitochondrial membranes and release of proteins
responsible for apoptosis
55.
Calcium influx andloss of ca homeostatis
• Mechanisms of cell damage
1. ATP depletion due to formation of permeability transition
pore following Ca2+ influx
2. Calcium mediated activation of phospholipases, proteases,
endonucleases and ATPases
3. Direct induction of apoptosis by activating caspases and
increasing mitochondrial permeability
Accumulation of Oxygenderived free radicles
• Common mechanism in chemical injuries, radiation, ischaemia-
reperfusion, cellular aging, bacterial phagocytois by leucocytes
• Commonest is reactive oxygen species, ROS
• Superoxide anion (O.
2)
• Hydrogen peroxide (H2O2)
• Hydroxyl ions (OH-
)
• ROS are a product of normal respiration and are removed by
cellular defence systems
• Increased production and reduced scavenging results in an marked
increase and potentiates cell injury
58.
Free radicles
• Mechanismof cell injury
• 1. Lipid peroxidation in membranes – ROS attack unsatuated fatty
acid membranes to yield unstable,reactive products
• 2. Oxidative modification of proteins –disrupts the conformation
of structural proteins
• 3. Lesions in DNA : breakages, crosslinking etc
Defects in membranepermeability
• Lead to loss of selectivity in permeability
• Mechanisms
• Reactive oxygen species (through lipid peroxidation)
• Decreased phospholipid synthesis –due to defective
mitochondria/hypoxia
• Increased phospholipid breakdown- due to activation of
phospholipases
• Cytoskeleton abnormalities
61.
DNA DAMAGE
• DNAcarries genetic instructions related to the growth, the development,
functioning and reproduction of a cell and a whole organism by extension
• Mutations (alterations in the genetic sequence) may occur; are inherited or
acquired
• Most mutations are identified and corected by the cell machinery through a process
known as proofreading
• Some mistakes may escape this step leading to a permanent alteration in fhe DNA
• This may result in formation of abnormal or suboptimally fuctioning proteins since
the DNA is the template upon which all proteins are made
• This may lead to varous genetic disorders disorders due to the absence or presence
of certain proteins
62.
CAUSES OF MUTATIONS
•Chemicals-nitrous acid, alkylating agents
• Exposure to radiation-x-rays cause DNA fragmentation,uv light
causes thymidine dimerization.
• Certain viruses
• Drugs
• Reactive oxygen species, ROS
• Metals- arsenic, nickel, cadnium
63.
Disorders of ribosomes
•May be due to abnormal biogenesis or abnormal function (improper protein
folding)
• Gene mutations affecting various ribosomal proteins result in abnormal
biogenesis of ribosomes
• Examples of conditions that may be caused by abnormal ribosomal proteins
1. Treacher-Collin Syndrome (TCS) _mutation in the gene coding for TCOF1
protein resultimg in a shorter/truncated protein; results in craniofacial
abnormalitie
2. Alopecia, neurological defects and endocinopathy syndrome (ANE
Syndrome); Mutation in RBM28 gene( required for the formatoon of 60S
subunit of ribosomes)
3. Prader Willi Syndrome(PWS)
4. Diamond Blackfan Aneamia (DBA)
5. Primary open angle Glaucoma (POAG)
64.
ER-GOLGI COMPLEX DISORDERS
•Correctly folded proteins leave transport vesicles that bud from the
ER to the ER-Golgi intermediate compartment
• Later delivered to the Golgi apparatus by vesicles or tubules
• Those that are improperly folded accumulate in the ER
• Functional defects may occur at the various steps of transport and
sorting within the ER and between the ER and the Golgi apparatus
• Example: Mutations affecting signal sequence affecting membrane
translocation or processing of signals may lead to primary
hypothyroidism, deficiency of coagulation factor X, Criggler Najjer
Syndrome II etc
Lysosomal storage diseases
•Lysosomes have more than 40 enzymes working on varied subtrate s.a RNA, DNA, complex
carbohydrates, phosphate esters etc
• If enzyme is congenitally absent (eg due to a genetic mutation), they become engorged with
subtrate to be broken down
Disease Deficient enzyme Substrate that accumulates
Fabry’s Disease Alpha galactosidase Glycolipids;glycoproteins
Tay sacchs Disease Hexosaminidase Gangliosides
Gaucher’s Disease Beta galactocerebrosidase Glucocerebroside
67.
Membrane damage
• Consequences
•Mitochondrial membranene – ATP depletion and apoptosis
• Plasma membrane – loss of osmotic balance and influx of fluids
• Lysosomal membranes – leakage of enzymes into the cytoplasm; cell
death by necrosis
69.
CYTOSKELETON DISORDERS
• Cytoskeletonmaintains cell shape & gives support to cell.
• 3types:Microfilaments,Microtubules,Intermediate filaments(6
subtypes).
Microfilaments disorders:
• Wiskott-aldrich syndrome.
• Congenital myopathies.(nemaline; myotubular; central core)
Microtubule disorders (tauopathies):
Hyperphosphorylation of tau protein due to mutation is the main
pathology in all disorders; neuroglial cells dysfunction; dementia
1. Alzheimer’s disease.
2. Pick’s disease.
3. Progressive supranuclear palsy.
4. Corticobasal degeneration.
70.
Cytoskeleton cont...
• Ciliadysfunction syndrome
1. Immotile cilia syndrome
2. Kartageners syndrome
3. Infertility syndrome
Intermediate filaments disorders:
Type 1&2 disorders
4. Epidermolysis bullosa
5. Keratoderma disorders
6. Hair & nail disorders
7. Other systems involvement
Type 3 disorders
8. Desminopathies(desmin)
9. Amyotropic lateral sclerosis(peripherin)
10. Alexander disease(GFAP)
11. Autosomal dominant juvenile cataract(vimentin)
WISKOTT-ALDRICH SYNDROME
• WASP(wiskottaldrich syndrome protein)
• Expressed in WBC & megakaryocytes& involved in reorganization of actin cytoskeleton.
• WASP is essential for function of T cells & platelets.
• Pt’s with WAS has defective gene in short arm of X chromosome(Xp11.23)
• Causes mutations in WASP leading to inability of actin to reorganize
• CD3 cannot be presented,T cells not activated,so B cells not activated,only IgM Ab’s are
produced.
• These defective cells also have problems with cell motility,difficulty attaching to other
cells.
• Pt presents as eczema, recurrent infections, microthrombocytopaenia, nose bleeds,
bloody diarrhea, auto immune disorders etc.
• It is a X linked recessive genetic trait.
73.
NEMALINE MYOPATHY
• Alsocalled rod myopathy or nemaline rod myopathy.
• Congenital hereditary neuromuscular disorder.
• Mutatons affecting 6 genes important in providing instructions for
producing proteins in muscle sarcomeres that are necessary for muscle
contraction.
ACTA1-alpha actin 1 gene, TPM3-alpha tropomyosin; NEB-nebulin gene.
TPM2-tropomyosin2; NNT1-troponin T1;CFL2-coffilin2 gene.
• So the proteins are disorganized,can’t interact normally & disrupts the
muscle contraction.
• Pt presents as muscle weakness arms,legs,trunk,throat,face
muscles,delayed motor development also seen.
Cell messaging /SIGNALTRANSDUCTION
DISORDERS
• Diabetes mellitus type 2 :
• Insulin reistance is one of tbe underlying mechanisms
• Studies suggest defects at post receptor level of insulin signalling involving
insulin receptor/substrate (IRS)-1 phosphatidylinositol (PI)3-Kinase pathway
• Results in dimished glucose uptake and utilization in insulin target tissues
• In pseudohypoparathyroidism (PHP) ;Hypocalcemia and
hyperphosphatemia due to parathyroid hormone insensitivity
rather than deficiency;
• Studies show that PTH fails to activate cAMP (second messenger) in
renal cortical tubular tissue
77.
REFERENCES
Robbins & cotrontextbook of pathology-8th
edition.
Anderson textbook of patholog
Review of physiology by Ganong
Endoplasmic reticulum storage diseases, Jonas Rutishauser, Martin
Spiess Swiss med wkly 2002; 132:211-222 www.smw.ch
NUCLEUS
• The nucleusis the largest cellular organelle.
• It is basically the control centre of the cell and harbours large
quantities of DNA.
• The nucleolus is the nuclear subdomain that assembles ribosomal
subunits, harbouring large amounts of RNA and proteins types found
in ribosomes
82.
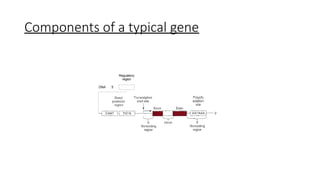
1.DNA
• Helical, double-strandedstructure
• Carries genetic material which control cell function by determining
which substances are synthesized within the cell- structures, enzyme,
chemicals.
• A gene is a sequence of DNA or RNA that codes for a molecule that
has a specific function.
83.
• The basicchemical compounds involved in the formation of DNA include:
• Phosphoric acid,
• A sugar called deoxyribose,
• four nitrogenous bases:
• Two purines - adenine and guanine
• Two pyrimidines -cytosine and thymine
Nucleotides;
• 1st
stage inDNA synthesis is the combination of one molecule of
phosphoric acid, one molecule of deoxyribose, and one of the four bases
to form an acidic nucleotide;
• deoxyadenylic, deoxythymidylic, deoxyguanylic, and deoxycytidylic acids
• Multiple numbers of nucleotides are then bound together to form two
strands of DNA
87.
• the strandsare usually bound together via weak hydrogen bonds
between the purine and pyrimidine bases such that;
• Each purine base adenine of one strand always bonds with a pyrimidine base
thymine of the other strand
• Each purine base guanine always bonds with a pyrimidine base cytosine.
88.
Genetic code;
• Whenthe two strands of a DNA molecule are split apart, this exposes the
purine and pyrimidine bases projecting to the side of each DNA strand,
these projecting bases form the genetic code.
• Usually consists of successive “triplets” of bases i.e each three successive
bases is a code word.
• The successive triplets eventually control the sequence of amino acids in
a protein molecule that is to be synthesized in the cell e.g proline, serine
89.
2.RNA
• Single stranded
•Because the DNA is located in the nucleus of the cell, yet most of the
functions of the cell are carried out in the cytoplasm, there must be
some means for the DNA genes of the nucleus to control the chemical
reactions of the cytoplasm, this is achieved by the RNA
• The code in the DNA is transferred to the RNA through the process of
transcription
90.
• The RNA,in turn, diffuses from the nucleus through nuclear pores into
the cytoplasmic compartment, where it controls protein synthesis.
• Building blocks of RNA;
• Ribose sugar
• Phosphoric acid
• Nitrogenous bases-adenine,guanine,cytosine,uracil(in place of
thymine in DNA)
91.
Types of RNA
1.Messenger RNA-which carries the genetic code to the cytoplasm for
controlling the type of protein formed.
2. Transfer RNA-which transports activated amino acids to the ribosomes
to be used in assembling the protein molecule.
3. Ribosomal RNA, which, along with about 75 different proteins, forms
ribosomes, the physical and chemical structures on which protein
molecules are actually assembled.
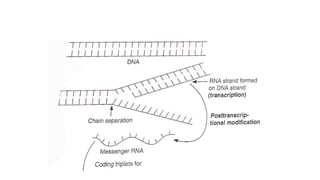
TRANSCRIPTION
• Process ofcreating an equivalent RNA copy of a sequence of DNA.
• During this process a DNA sequence is read by DNA polymerase, which
produces a complementary , antiparallel strand.
• Thymine (T) in the DNA segment is usually replaced by Uracil(U) in the
complementary RNA strand
94.
• The stretchof DNA transcribed into an RNA molecule is called a transcription
unit and encodes atleast one gene
• If the gene transcribed encodes for a protein, the result of transcription is
mRNA, which will be used to create that protein via translation
• A DNA transcription unit encoding for a protein contains not only the
sequence that will eventually be directly translated into the proteins,
regulatory sequences that direct and regulate synthesis of that protein
Initiation
• RNA polymeraseidentifies and attaches to promoter region( a sequence
of nucleotides immediately ahead of the initial gene)
• The RNA polymerase has an appropriate complementary structure that
recognizes this promoter and becomes attached to it.
• Essential for initial formation of RNA molecule
97.
Elongation
• After theRNA polymerase attaches to the promoter, the polymerase
causes unwinding of about two turns of the DNA helix and separation
of the unwound portions of the two strands.
98.
• Then thepolymerase moves along the DNA strand, temporarily
unwinding and separating the two DNA strands at each stage of its
movement. As it moves along, it adds at each stage a new activated
RNA nucleotide to the end of the newly forming RNA chain
99.
termination
• When theRNA polymerase reaches the end of the DNA gene, it
encounters a new sequence of DNA nucleotides called the chain-
terminating sequence; this causes the polymerase and the newly formed
RNA chain to break away from the DNA strand. Then the polymerase can
be used again and again to form still more new RNA chains.
102.
Post transcription modification
•After transcription, editing is done to make the RNA functional(basically
converting the pre mRNA to a mature mRNA
• Introns- non functional segments of DNA that are removed
• Exons- segments of DNA coding for proteins are then rejoined by the enzyme
ligase
• A guanine triphosphatase cap is added to the 5’ end of the newly copied mRNA
• A poly A tail is added to the 3’ end of the RNA
103.
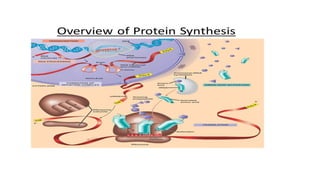
TRANSLATION
• This isthe first process of protein biosynthesis, and involves production
of proteins by decoding m RNA produced through transcription
• Each codon decoded guides the formation of a specific
polypeptide/protein
• When a molecule of messenger RNA comes in contact with a ribosome, it
travels through the ribosome, beginning at a predetermined end of the
RNA molecule specified by an appropriate sequence of RNA bases called
the “chain-initiating” codon.
104.
• While themessenger RNA travels through the ribosome, a protein
molecule is formed-translation
• when a “stop” (or “chain-terminating”) codon slips past the ribosome,
the end of a protein molecule is signaled and the protein molecule is
freed into the cytoplasm.
• Ribosomes consist of two parts, a large subunit and a small subunit. They
contain a binding site for mRNA and two binding sites for transfer RNA
(tRNA) located in the large ribosomal subunit
Activation
• Each aminoacid is activated by a chemical process in which ATP
combines with the amino acid to form an adenosine monophosphate
complex with the amino acid (AMP-AA complex) giving up two high-
energy phosphate bonds in the process.
• The activated amino acid, having an excess of energy, then combines
with its specific transfer RNA to form an amino acid–tRNA complex( AA-
tRNA ),and at the same time, releases the adenosine monophosphate
(AMP)
107.
• The activatedamino acid, having an excess of energy, then combines
with its specific transfer RNA to form an amino acid–tRNA complex and,
at the same time, releases the adenosine monophosphate.
108.
Initiation
• The transferRNA carrying the amino acid then comes in contact with the
messenger RNA molecule in the ribosome- typically starts at the start
codon AUG
• The anticodon of the transfer RNA attaches temporarily to its specific
codon of the messenger RNA, lining up the amino acid in appropriate
sequence to form a protein molecule
109.
Elongation
• Peptide bondsare formed between the successive amino acids, thus
adding progressively to the protein chain.
• This is under the influence of the enzyme peptidyl transferase.
• These chemical events require energy from two additional high-energy
phosphate bonds, making a total of four high-energy bonds used for
each amino acid added to the protein chain.
110.
Termination
• Translation continuesuntil the stop codon(AUG) is reached , and the
polypeptide chain is released.
• The tRNA molecules are used again
• The mRNA molecules are also reused approximately 10 times beifore
being replaced.
112.
Post Translational Modification
•After the polypeptide chain is formed, it is modified to the final protein
by:
a) One or more of a combination of reactions that include hydroxylation,
carboxylation, glycosylation, or phosphorylation of amino acid residues
b) cleavage of peptide bonds that converts a larger polypeptide to a smaller form.
c) folding and packaging of the protein into its ultimate, often complex
configuration.
PRESENTATION OUTLINE
• SUMMARYOF PROTEIN SYNTHESIS
• ABNORMALITIES
• CLASSIFICATION
• DISCUSS IN SUMMARY ONE DISORDER IN EACH CLASS
• DIAGNOSIS
116.
MUTATIONS
Mutations are changesin the nucleotide sequence of DNA.
Can either be genetic/hereditary or acquired/somatic
A hereditary mutation is a mistake that is present in the DNA of
virtually all body cells.
Many mutations are repaired by enzymes
117.
CAUSES OF MUTATIONS
Spontaneousmutations
Caused during DNA replication/incorporation of incorrect
nucleotide into growing DNA chain
Induced mutation
Changes in DNA brought about by some environmental
factors eg mutagens such as
Chemicals-nitrous oxide, alkylating agents
Smoking,
Exposure to radiation
Drugs-cause cross linkage of DNA thus blocking replication
118.
TYPES OF MUTATIONS
Classifiedinto:
genome mutation: (whole chromosome)
Gene mutations-change in the nucleotide sequence of a gene
May only involve a single nucleotide
Chromosomal mutation-issue with chromosome structure
Loss or gain of a part of a chromosome.
119.
TYPES OF GENEMUTATIONS
• Point mutation-single nucleotide base may be substituted by a
different base.
• Insertion-one/two base pairs may be inserted into the dna
• Deletion-one/two base pairs may be deleted from the dna
• Both insertion and deletion mutations lead to alterations in the
reading frame of the DNA strand hence known as frame shift
mutations.
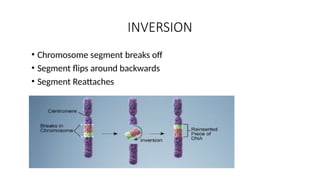
TRANSLOCATION
• Involves twochromosomes that aren’t homologous
• Part of one chromosome transferred to another chromosomes
125.
Nondisjunction
• Failure ofa chromosome to separate during meiosis
• Causes gamete to have too many or too few
chromosomes
• Disorders:
• Klinefelter’s Syndrome- XXY chromosomes
Down Syndrome- three 21st
chromosomes
Turner Syndrome- single X chromosome
126.
END RESULTS
• Interfereswith protein synthesis-causes loss of function
• Suppress transcription with gene deletions and point
mutations involving promoter sequence
• Produce abnormal mRNA from mutations affecting introns
• Defects are carried to translation. Translation is affected if
a stop codon is created within an exon
• Abnormal proteins without impairing any step in protein
synthesis
127.
GENETIC DISORDERS
• Polygenic/multifactorialinheritance-mutation caused by
both genetic and environmental factors e.g. HTN,DM
• Monogenic/mendelian disorders-resulting from single gene
mutations. They follow mendelian factor of inheritance .
• Chromosomal disorders- are associated with numerical
/structural changes in chromosomes
• Mitochondrial –caused by a mutation in the non
chromosomal DNA of the mitochondria
128.
MONOGENIC DISORDER
• Resultfrom mutations in the DNA sequence of single
genes.
• As a result the protein the gene codes for is either altered
or missing
• Gene expression is usually described as dominant or
recessive
a. Autosomal dominant
b. Autosomal recessive
c. Sex linked (recessive) involving x chromosome
129.
AUTOSOMAL DOMINANT
• Manifestedin the heterozygous state
• One parent of an index case is usually affected
• Both males and females are affected
• Both can transmit the condition
• Clinical features can be modified by reduces penetrance
and variable expressivity
• Reduced penetrance-some individuals inherit the mutant
gene but are phenotypically normal
• Variable expressivity-mutant gene is expressed differently
among individuals
AUTOSOMAL RECESSIVE
• Resultwhen both alleles at a given locus are mutants
• Feature include
• The trait does not usually affect the arents but the siblings
may show dx
• Siblings have 1 chance in 4 of being affected
• Expression of the defect tends to be more uniform than in
AD disorders
• Complete penetrance is common
• Onset is frequently early in life
SEX LINKED X
•Sex linked traits are x linked
• Can be dominant or recessive
• No y linked inheritance since all genes encoded in the male specific
region of y are related to spermatogenesis.males with mutations
affecting y linked genes are usually infertile.
• An affected male does not transmit the disorder to his sons but all
daughters are carriers
134.
X LINKED DISORDERS
•Muskuloskeletal-duchenne muscular dystrophy
• Blood-hemophilia A and B
• Immune-agammaglobulinemia,wiskott-Aldrich syndrome
• Metabolic-diabetes insipidus,lesch-nyhan syndrome
• Nervous-fragile x syndrome
135.
POLYGENIC INHERITANCE
• Combinedaction of environmental influences and 2 or more mutant
genes
• Expression defers by number of genes
• Continous traits eg height vs discontinuous traits eg albinism
• Examples include
• Cleft lip/palate congenital heart disease
• Coronary heart disease hypertension
• Gout diabetes mellitus
• Pyloric stenosis
136.
CHROMOSOMAL GENETIC DISORDR
•In these disorders, entire chromosomes,
or large segments of them, are missing, duplicated, or
otherwise altered.
• organized into two groups:
1) Numerical Abnormalities: When an individual is
missing either a chromosome from a pair (monosomy) or
has more than two chromosomes of a pair (trisomy)
2) Structural Abnormalities: When the chromosome's
structure is altered
137.
• Could alsobe divided into:
Autosomes-are the first 22 homologous pairs of human
chromosomes that do not influence the sex of an
individual e.g. down syndrome, trisomy 13,trisomy 18
Sex chromosomes- e.g. the 23rd
pair of chromosomes
that determine the sex of an individual.
• Examples -klinefelter xxy,-extra sex chromosome
• turner xo-missing sex chromosome
138.
MITOCHONDRIAL INHERITANCE
• Maternalinheritance
• This is because the ova contain numerous mitochondria
within their abundant cytoplasm
• Spermatozoa contain few
• Diseases-mitochondrial myopathies e.g. chronic
progressive external opthamoplegia ,myoclonic epilepsy
with ragged fibers
139.
DIAGNOSIS OF GENETICDISORDERS
Hx,physical exam,labworks and imaging
There are several ways to determine whether a child will
have a genetic disorder
Two main ways to diagnose:
1.Analysis of fetal cells
• Amniocentesis
• Chorionic villus biopsy
2.Imaging techniques
• Ultrasonography (computerized image)
• Fetoscopy (direct observation)
140.
SUMMARY OF PROTEINSYNTHESIS
DISORDERS
• CHROMOSOMAL DISORDERS
• MULTIGENE DISODERS
• SINGLE GENE DISORDERS
142.
REFERENCE
• ROBBINS ANDCOTRAN,PATHOLOC BASIS OF DISEASE 7TH
EDITION
• GANONGS REVIEW OF MEDICAL PHYSIOLOGY 25TH
EDITION
• TEXTBOOK OF MEDICAL PHYSIOLOGY GUYTON AND HALL.