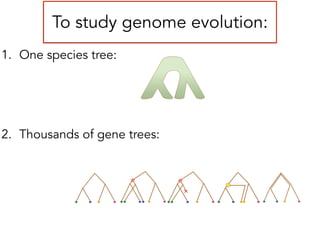
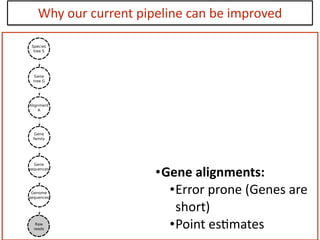
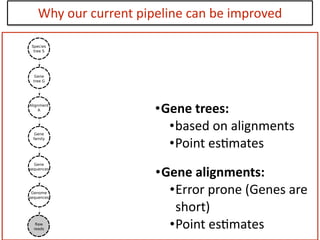
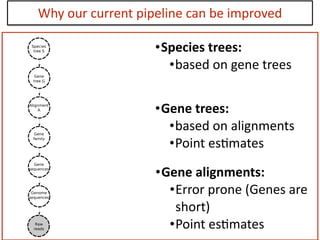
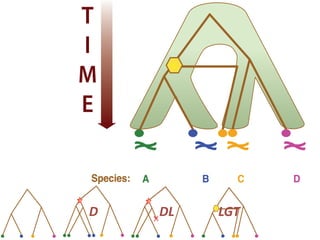
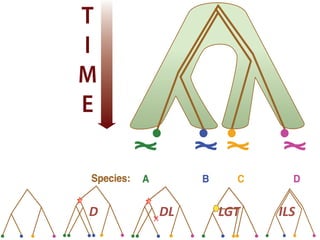
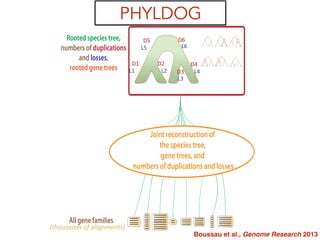
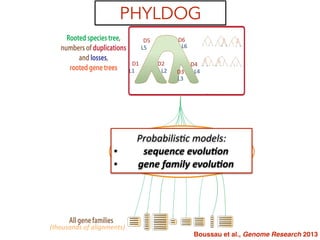
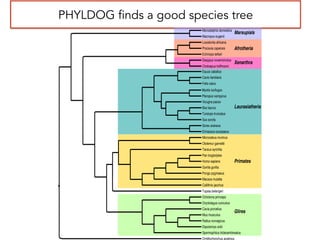
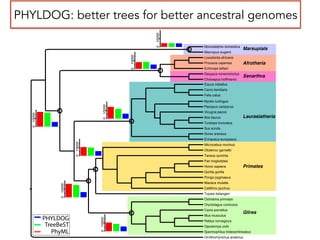
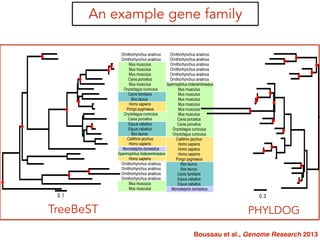
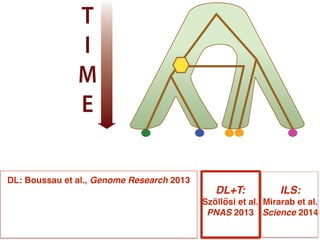
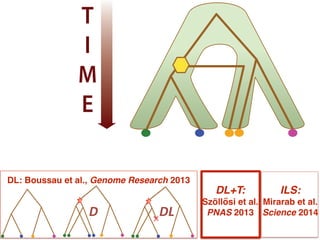
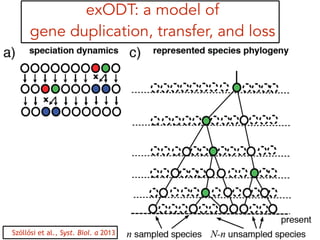
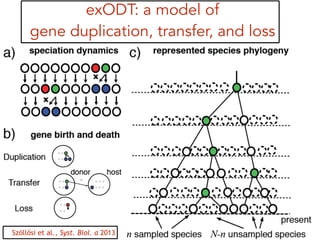
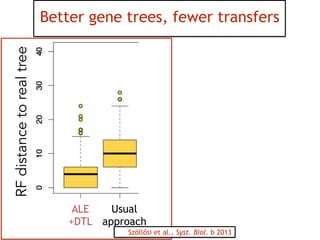
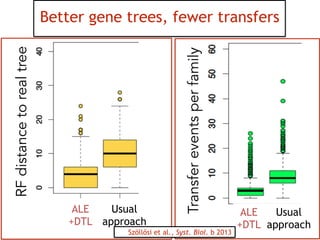
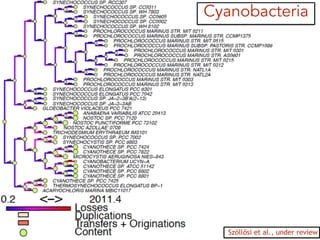
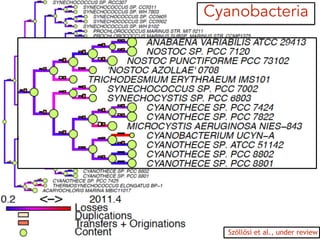
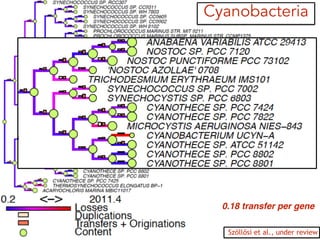
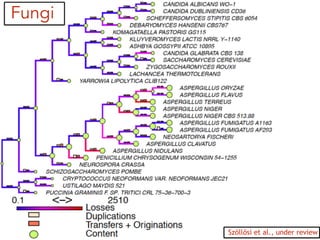
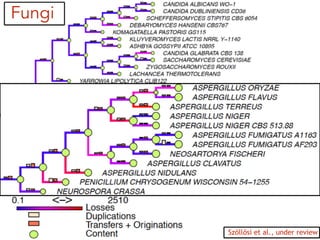
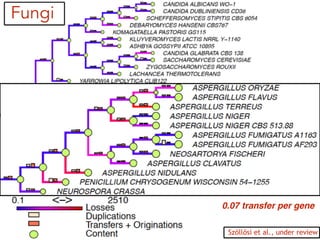
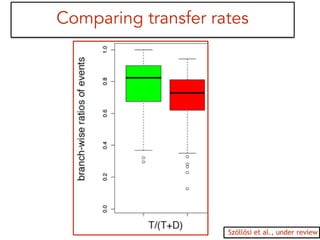
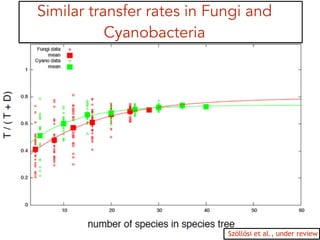
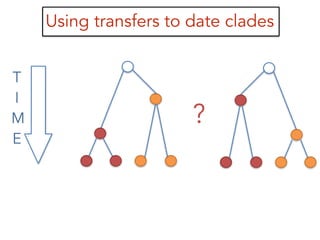
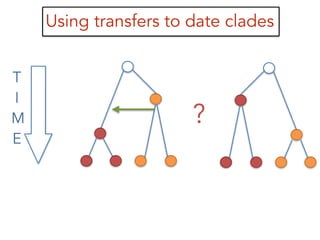
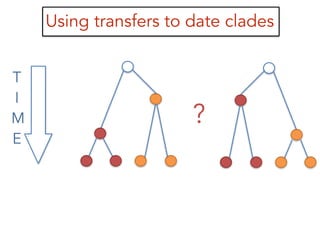
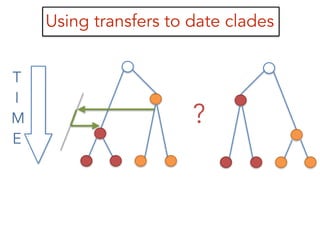
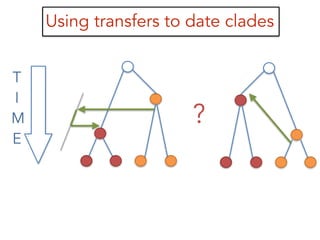
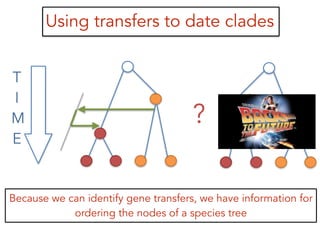
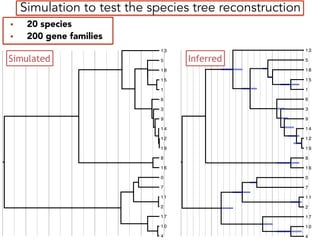
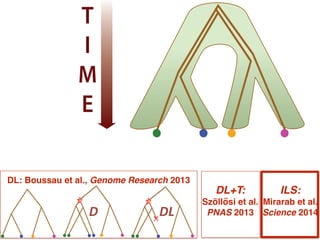
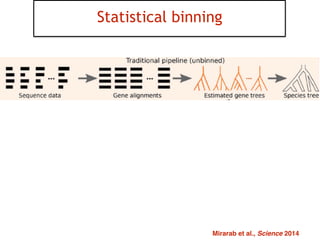
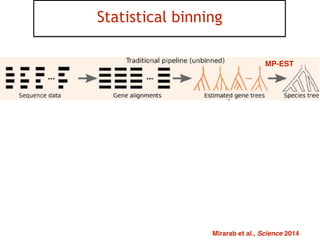
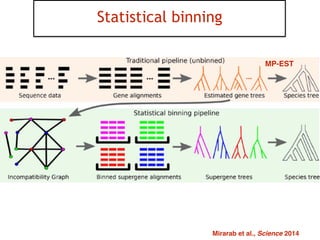
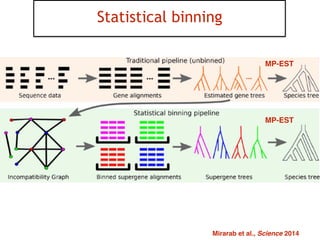
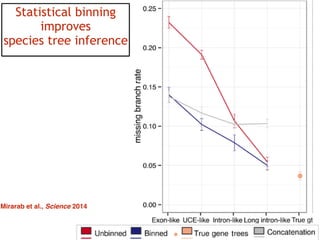
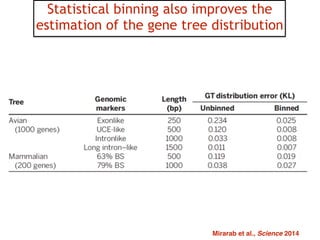
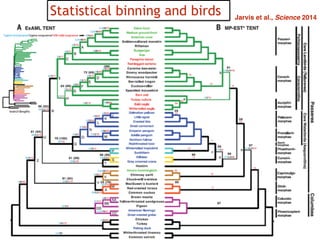
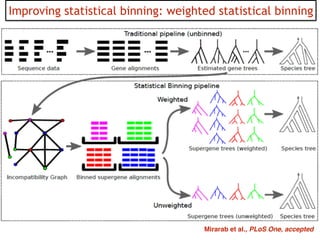
This document discusses the use of models like Phyldog and Exodt to study gene duplication, transfer, and loss, thereby enhancing our understanding of genome evolution in various organisms, including mammals, fungi, and cyanobacteria. It outlines the collaborations involved, the plans for research focusing on different approaches to gene analysis, and improvements made to existing algorithms for more accurate phylogenetic trees. The document also highlights the age and gene family composition of the species studied and the expectation of transfer frequencies among them.







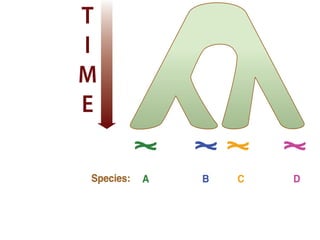
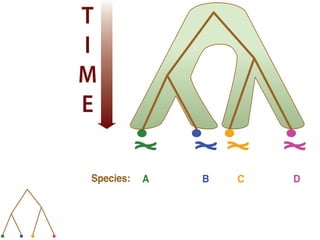
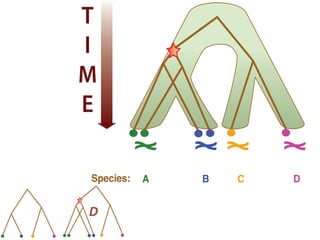
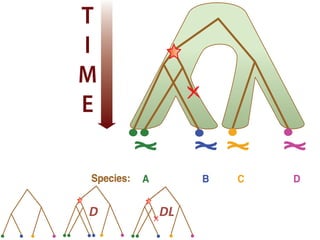









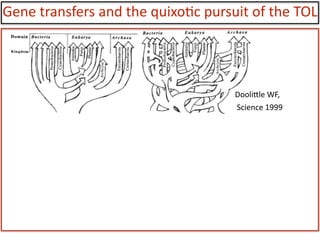
![Gene
transfers
and
the
quixo:c
pursuit
of
the
TOL
DooliYle
WF,
Science
1999
“The monistic concept of a single universal tree appears […]
increasingly obsolete. […][It is] no longer the most
scientifically productive position to hold[…][It] accounts for
only a minority of observations from genomes.”!
Bapteste, O’Malley, Beiko, Ereshefsky, Gogarten, Franklin-Hall,
Lapointe, Dupré, Dagan, Boucher, Martin, !
Biology Direct 2009.](https://image.slidesharecdn.com/bastienboussauucl2015-150610082027-lva1-app6891/85/Models-of-gene-duplication-transfer-and-loss-to-study-genome-evolution-35-320.jpg)





































































![谷歌留痕技术 [ 𝙩𝙤𝙥 𝟮𝟯𝟯. 𝙘 𝙤𝙢 ]](https://cdn.slidesharecdn.com/ss_thumbnails/top233-260130174328-3833018c-thumbnail.jpg?width=640&height=640&fit=bounds)







