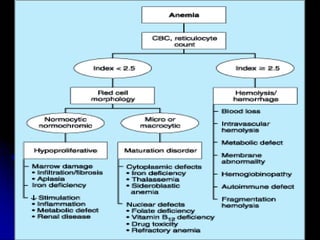
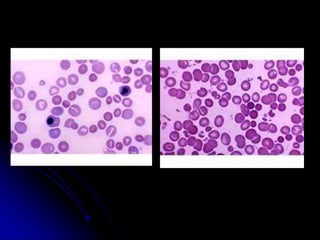
1. A 19-year-old man presented with symptoms of anemia including pallor, weakness and giddiness. Laboratory tests revealed severe normochromic normocytic anemia.
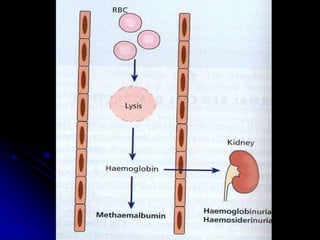
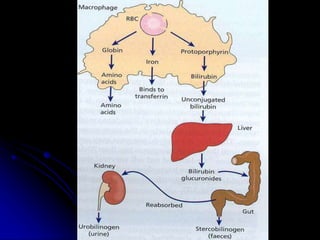
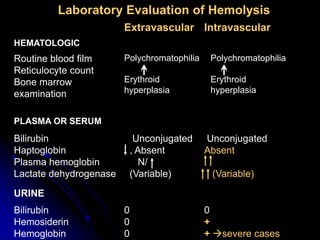
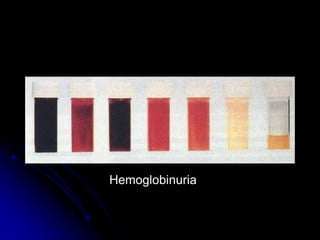
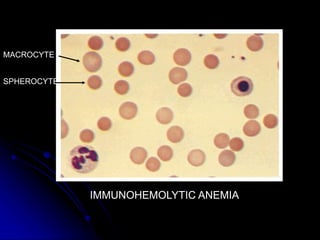
2. Further investigation showed evidence of hemolysis including reticulocytosis, elevated bilirubin and LDH, and low haptoglobin. The direct antiglobulin test was positive, indicating autoimmune hemolytic anemia.
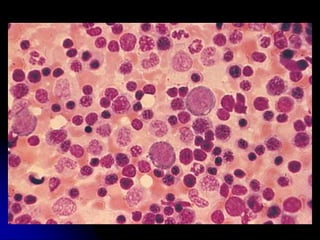
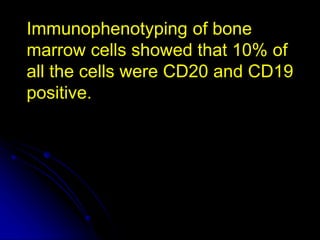
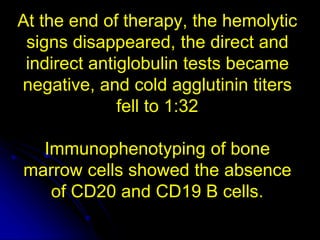
3. The patient was initially treated with steroids which improved his symptoms and lab values. However, upon tapering steroids, his anemia relapsed. Additional testing found CD20+ B cells in his bone marrow. Rituximab treatment eliminated the B cells and induced






































































![CTEV [ clubfoot] DR ARUN LAL ,DR MOHAMED ASHRAF travancore medical college k...](https://cdn.slidesharecdn.com/ss_thumbnails/ctevclubfootdrarunlaldrmohamedashraftravancoremedicalcollegekollamkeralaindia-260208063247-18fc466c-thumbnail.jpg?width=640&height=640&fit=bounds)











![PERI-PROSTHETIC FRACTURE NAIL-PLATE CONSTRUCT [NPC].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/drarunkumardrmohamedashrafperiprostheticfrasturenail-plateconstructnpc-260209164459-7e9d15a1-thumbnail.jpg?width=640&height=640&fit=bounds)





