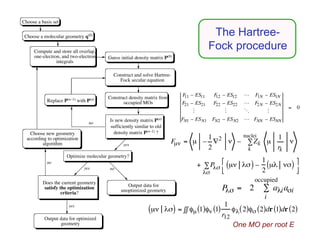
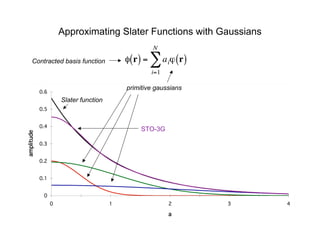
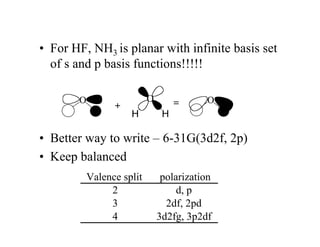
The document discusses basis sets and linear combination of atomic orbitals (LCAO) wave functions used in Hartree-Fock theory. It explains that atomic orbitals like Slater-type orbitals are approximated using Gaussian functions for calculations. Basis sets like STO-3G use a minimal number of primitive Gaussians to represent atomic orbitals, while more advanced basis sets like 6-31G add polarization and diffuse functions for improved accuracy.