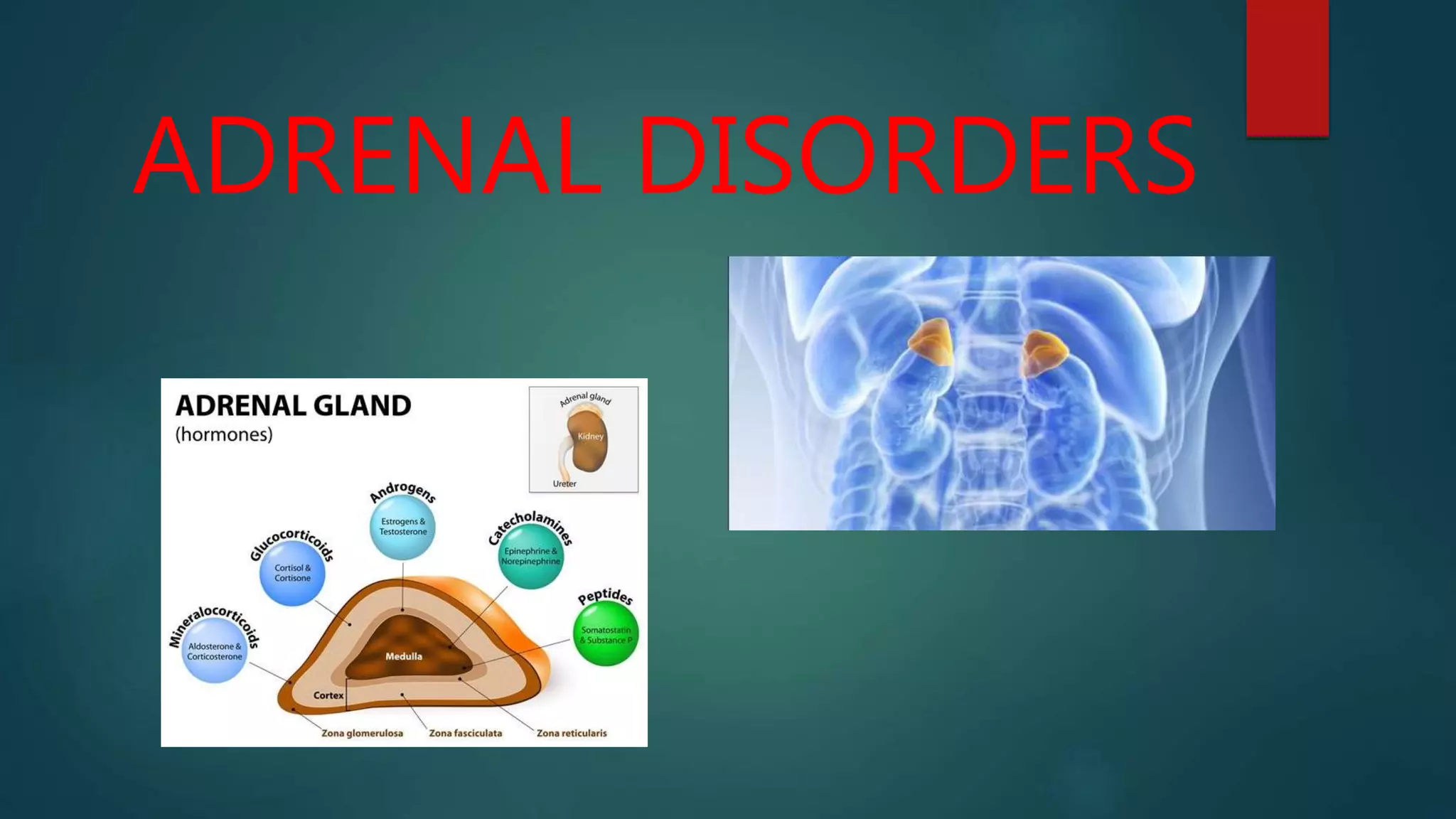
- Adrenal disorders include primary hypoadrenalism (Addison's disease), congenital adrenal hyperplasia, primary hyperaldosteronism, and pheochromocytoma.
- Addison's disease is caused by destruction of the adrenal cortex leading to deficiencies in glucocorticoids, mineralocorticoids, and sex steroids. Common causes include autoimmune disease and tuberculosis.
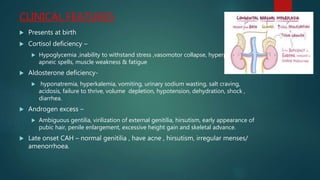
- Congenital adrenal hyperplasia is caused by enzyme deficiencies leading to cortisol deficiency and excess androgen production, often causing virilization in females.
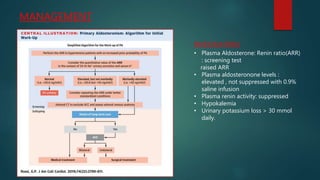
- Primary hyperaldosteronism is characterized by excessive aldosterone secretion causing hypokalemia and hypertension.