Arteriosclerosis
▪Hardening and thickeningof arterial wall
▪Loss of elasticity
▪3 general patterns:
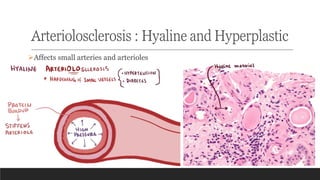
1. Arteriolosclerosis- affects small arteries and arterioles; hyaline and
hyperplastic
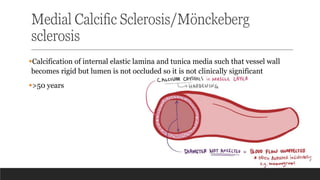
2. Mönckeberg sclerosis/ medial calcific sclerosis – calcification of tunica media
and internal elastic membrane; lumen not occluded
3. Atherosclerosis- clinically most significant pattern
3.
Atherosclerosis
▪Greek "athero" meaninggruel or wax (necrotic core area at the core) of the
atherosclerotic plaque and "sclerosis" for hardening referring (fibrous cap)
▪Chronic inflammatory disorder of medium and large arteries
▪Atherosclerosis is a potentially serious condition where arteries become
clogged with fatty substances called plaques, or atheroma.
▪Is the underlying pathogenesis behind the coronary, cerebral and peripheral vascular
disease
▪Causes more mortality and morbidity in the developed world than any other
disorders(manifested as death by MI, stroke, etc.)
4.
Epidemiology
▪CAD remains theleading cause of death in the western world
▪Although atherosclerosis-associated IHD is widespread among developed
nations, risk reduction and improved therapies have combined to moderate the
associated mortality
▪Reduced mortality from infectious diseases and the adoption of Western
lifestyles has led to increased prevalence of IHD in developing nations
6.
Risk factors
▪Prevalence andseverity of atherosclerosis and
IHD among individuals and groups are related to
number of risk factors
▪The effect of risk factors can be multiplicative
rather than additive
▪People with a combination of risk factors are at
greatest risk
7.
Constitutional/non-modifiable risk factors
Genetics
▪Familyhistory- most important independent risk factor
▪usually polygenic, relating to familial clustering of other established risk factors,
such as hypertension or diabetes
▪monozygotic twin of an affected individual has an eightfold increased risk and a
dizygotic twin a fourfold increased risk of dying from CAD
Age
▪clinical manifestations evident usually at the middle age when lesion cross a
critical threshold
8.
Non-modifiable risk factors
Sex
▪Premenopausalwomen usually protective against atherosclerosis and its
complications
▪In postmenopausal women the incidence increases and at older ages is greater
than in men
African-American descent
▪Genetics- carry gene that makes them more sensitive to salt which can lead to
HTN
▪lifestyle
9.
Modifiable risk factors
Dyslipidemia(specifically hypercholesteremia)
▪Sufficient to initiate lesion development even in the absence of other risk factors
▪Increased LDL, decreased HDL
▪Exercise and moderate consumption of ethanol raise HDL levels whereas obesity
and smoking lowers it
▪Statins
10.
Modifiable risk factors
Hypertension
▪canincrease the risk of IHD by approximately 60% versus normotensive
populations
▪Chronic hypertension is the most common cause of left ventricular hypertrophy
Smoking
▪Prolonged (years) smoking of one pack of cigarettes or more daily doubles the
death rate from IHD
▪Smoking cessation reduces that risk substantially
12.
Modifiable risk factors
Physicalactivity
▪Regular exercise has a protective effect, whereas inactivity roughly doubles the
risk of CAD and is a major risk factor for stroke.
Obesity
Alcohol consumption
Social deprivation
13.
Modifiable risk factors
Diabetesmellitus
▪induces hypercholesterolemia and markedly increases the risk of atherosclerosis
▪Twice likely to have MI than those without DM
▪Increased risk of stroke
▪100 times increased risk of atherosclerosis-induced gangrene in lower
extremities
15.
Additional risk factors
About20% of all cardiovascular events occur in the absence of overt risk factors (e.g.,
hypertension, hyperlipidemia, smoking, or diabetes)
Inflammation
▪Present during all stages of atherosclerosis and is linked with plaque formation and
rupture
C-reactive protein (CRP)
▪Simplest to measure and most sensitive marker related to IHD
▪Acute phase protein synthesized by liver which augments the innate immune response
16.
Additional risk factors
▪PlasmaCRP - most important marker in MI, stroke, peripheral arterial disease and
sudden cardiac death
▪Useful marker for accessing the effect of risk reduction measures (decreased CRP)
Hyperhomocysteinemia
▪Homocystinuria (due to rare inborn errors of metabolism) results in elevated circulating
homocysteine (>100 μmol/L) and is associated with premature vascular disease
Hemostatic factors
▪Platelet activation and high plasma fibrinogen concentrations are associated with an
increased risk of coronary thrombosis
17.
Pathogenesis-Response to injurytheory
▪This model views atherosclerosis as a chronic inflammatory and healing
response of the arterial wall to endothelial injury
▪Lesion progression occurs through interaction of modified lipoproteins,
macrophages, and T lymphocytes with endothelial cells and smooth muscle cells
of the arterial wall
▪Atherosclerosis progresses according to the following scheme:
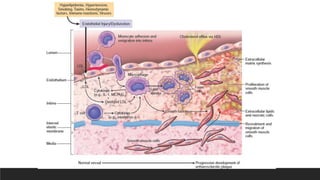
1. Endothelial injury and dysfunction causing increased vascular permeability,
leukocyte adhesion and thrombosis
2. Accumulation of lipoproteins (mainly oxidized LDL)in the vessel wall
18.
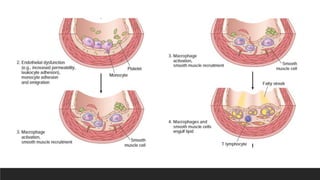
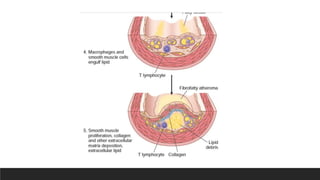
Pathogenesis-Response to injurytheory
3. Monocyte adhesion to the endothelium, followed by migration into the intima
and transformation into macrophages and foam cells
4. Platelet adhesion
5. Factor release from activated platelets, macrophages, and endothelial cells,
inducing smooth muscle cell recruitment from the media
6. Smooth muscle cell proliferation, extracellular matrix production, and
recruitment of T cells.
7. Lipid accumulation both extracellularly and within cells (macrophages and
smooth muscle cell)
20.
General overview ofpathogenesis
Endothelial injury due to various etiologic factors especially
hemodynamic factors and hyperlipidemia
Due to endothelial injury, LDL particles can leak into intimal layer
from circulation(lumen) where it gets oxidized
When LDL gets oxidized it becomes a pro-inflammatory antigen
that induces an immune response
21.
General overview ofpathogenesis
Inflammatory cells like monocytes come to fight this antigen and is
converted into macrophages after entering the intima
Macrophages engulf the oxidized LDL and form FOAM CELLS
Accumulation of foam cells underneath the endothelium creates fatty
streak (1st marker of atherosclerosis)
22.
General overview ofpathogenesis
Due to endothelial injury, platelets comes into action and adhere with
the endothelial cells
They release PDGF, FGF and TGF-α which stimulate smooth muscle
cells proliferation and promote their migration from tunica media to
intima
SMC then proliferate and stimulate the production of extracellular matrix
leading to the formation of PLAQUE (stable plaque)
24.
General overview ofpathogenesis
Over time foam cells undergo necrosis and release matrix
metalloproteinases (MMPs) which destroys the outer fibrous cap of
plaque
Now the atheroma is exposed to vessel lumen-Unstable plaque
Platelet forms fibrin clot and clot can occlude the lumen even more or
even can embolize
29.
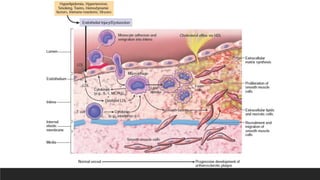
1. Endothelial injury
▪earlyhuman lesions begin at sites of morphologically intact endothelium
▪Non-denuding endothelial dysfunction underlies most human atherosclerosis;
the intact but dysfunctional endothelial cells exhibit increased endothelial
permeability, enhanced leukocyte adhesion and altered gene expression
▪2 most important causes are: hemodynamic disturbances and
hypercholesterolemia
30.
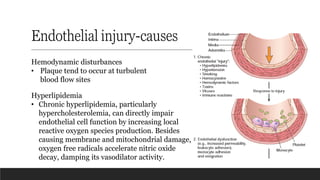
Endothelial injury-causes
Hemodynamic disturbances
•Plaque tend to occur at turbulent
blood flow sites
Hyperlipidemia
• Chronic hyperlipidemia, particularly
hypercholesterolemia, can directly impair
endothelial cell function by increasing local
reactive oxygen species production. Besides
causing membrane and mitochondrial damage,
oxygen free radicals accelerate nitric oxide
decay, damping its vasodilator activity.
31.
Inflammation
▪Triggered by theaccumulation of cholesterol crystals and free fatty acids
▪Oxidized LDL act as proinflammatory antigen that recruits leukocytes including
monocytes which converts into macrophages in the intima
▪Oxidized LDL is taken up by scavenger receptor in macrophages to transform it
into foam cell
▪Net result of macrophage and T cell activation is the local production of
cytokines and chemokines that recruit and activate more inflammatory cells
32.
Inflammation …
▪Activated macrophagesproduce ROS that enhance LDL oxidation, and elaborate
growth factors that drive smooth muscle cell proliferation
▪Activated T cells in the growing intimal lesions elaborate inflammatory cytokines
like interferon-γ, which, in turn, can activate macrophages, endothelial cells and
smooth muscle cells
▪These leukocytes and vascular wall cells release growth factors that promote
smooth muscle cell proliferation and synthesis of extracellular matrix proteins
▪Thus, chronic inflammation contributes to initiation and progression of
atherosclerotic lesions
34.
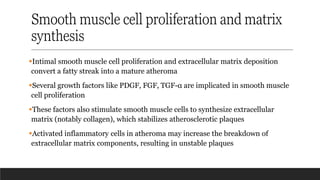
Smooth muscle cellproliferation and matrix
synthesis
▪Intimal smooth muscle cell proliferation and extracellular matrix deposition
convert a fatty streak into a mature atheroma
▪Several growth factors like PDGF, FGF, TGF-α are implicated in smooth muscle
cell proliferation
▪These factors also stimulate smooth muscle cells to synthesize extracellular
matrix (notably collagen), which stabilizes atherosclerotic plaques
▪Activated inflammatory cells in atheroma may increase the breakdown of
extracellular matrix components, resulting in unstable plaques
39.
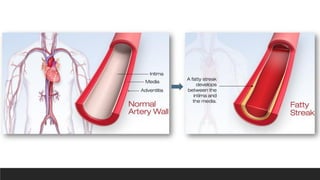
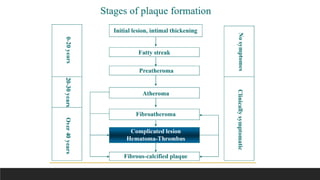
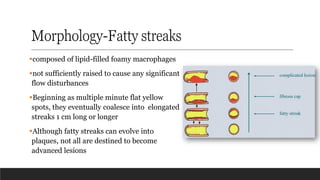
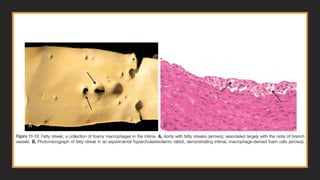
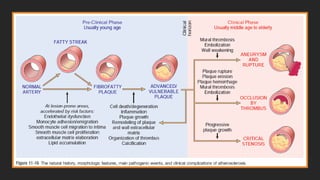
Morphology-Fatty streaks
▪composed oflipid-filled foamy macrophages
▪not sufficiently raised to cause any significant
flow disturbances
▪Beginning as multiple minute flat yellow
spots, they eventually coalesce into elongated
streaks 1 cm long or longer
▪Although fatty streaks can evolve into
plaques, not all are destined to become
advanced lesions
41.
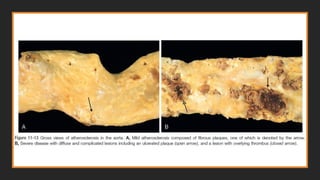
Morphology-Atherosclerotic plaque
GROSS
▪Atheromatous plaquesare white-yellow and encroach on the lumen of the artery
▪Superimposed thrombus over ulcerated plaques is red-brown
▪Plaques vary in size but can coalesce to form larger masses
▪Although focal and sparsely distributed at first, with time atherosclerotic lesions
can become larger, more numerous, and more broadly distributed
43.
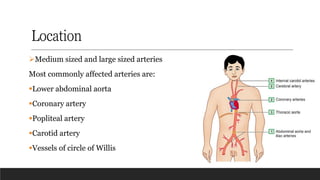
Location
➢Medium sized andlarge sized arteries
Most commonly affected arteries are:
▪Lower abdominal aorta
▪Coronary artery
▪Popliteal artery
▪Carotid artery
▪Vessels of circle of Willis
44.
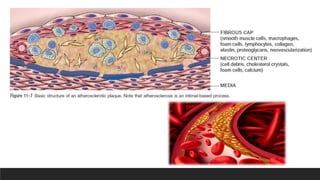
Microscopy-Atherosclerotic plaque
▪Atherosclerotic plaqueshave 3 principal components:
(1) smooth muscle cells, macrophages, and T cells
(2) extracellular matrix, including collagen, elastic fibers, and proteoglycans
(3) intracellular and extracellular lipid
▪Superficial fibrous cap composed of SMCs and dense collagen
▪To the side of the cap more cellular area with macrophages, T cells and SMCs
▪Deep to the fibrous cap is a necrotic core containing lipids, debris from dead
cells, foam cells, fibrin, variably organized thrombus and plasma proteins
47.
Microscopy-Atherosclerotic plaque
▪The peripheryof the lesions demonstrate neovascularization
▪Most atheromas contain abundant lipid, but some plaques (“fibrous plaques”) are
composed almost exclusively of smooth muscle cells and fibrous tissue.
▪Atherosclerotic plaques are susceptible to the following clinically important
pathologic changes:
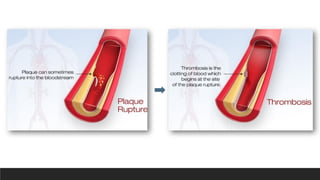
1. Rupture, ulceration, or erosion of the surface of atheromatous plaques
exposes highly thrombogenic substances and leads to thrombosis, which may
partially or completely occlude the vessel lumen
If the patient survives, the clot may become organized and incorporated into the
growing plaque.
50.
Microscopy-Atherosclerotic plaque
2. Hemorrhageinto the plaque
Rupture of the overlying fibrous cap, or of the thin-walled vessels in the areas of
neovascularization, can cause intraplaque hemorrhage; a contained hematoma
may expand the plaque or induce plaque rupture.
3. Atheroembolism: Plaque rupture can discharge atherosclerotic debris into
the bloodstream, producing microemboli.
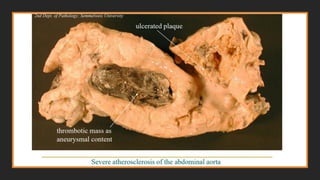
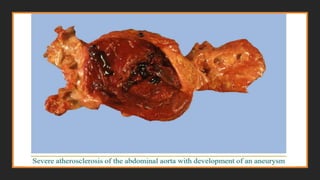
4. Aneurysm formation. Atherosclerosis-induced pressure or ischemic
atrophy of the underlying media, with loss of elastic tissue, causes weakness and
potential rupture.
52.
Consequences of AtheroscleroticDisease
Myocardial infarction (heart attack), cerebral infarction (stroke), aortic
aneurysms, and peripheral vascular disease (gangrene of the legs) are the
major consequences of atherosclerosis.
54.
Atherosclerotic stenosis
▪In smallarteries, atherosclerotic plaques can gradually occlude vessel lumen,
compromising blood flow and causing ischemic injury
▪Critical stenosis is the stage at which the occlusion is sufficiently severe to
produce tissue ischemia
▪In the coronary circulation, this typically occurs at when the occlusion produces a
70% decrease in luminal cross-sectional area
▪Consequences :Mesenteric occlusion and bowel ischemia, sudden cardiac death,
chronic IHD, ischemic encephalopathy and intermittent claudication
55.
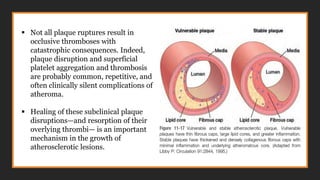
Acute plaque change
▪Plaquechanges falls in 3 general categories:
• Rupture/fissuring, exposing highly thrombogenic plaque constituents
• Erosion/ulceration, exposing the thrombogenic subendothelial basement
membrane to blood
• Hemorrhage into the atheroma, expanding its volume
▪Plaques containing large areas of foam cells and extracellular lipid, and those in
which the fibrous caps are thin or contain few SMCs or have clusters of
inflammatory cells, are more likely to rupture-“vulnerable plaques”
56.
▪ Not allplaque ruptures result in
occlusive thromboses with
catastrophic consequences. Indeed,
plaque disruption and superficial
platelet aggregation and thrombosis
are probably common, repetitive, and
often clinically silent complications of
atheroma.
▪ Healing of these subclinical plaque
disruptions—and resorption of their
overlying thrombi— is an important
mechanism in the growth of
atherosclerotic lesions.
57.
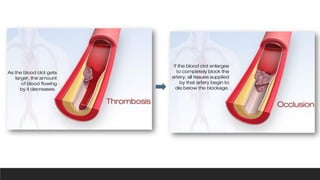
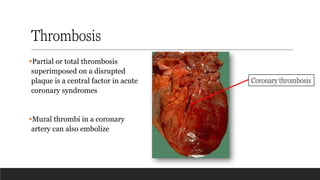
Thrombosis
▪Partial or totalthrombosis
superimposed on a disrupted
plaque is a central factor in acute
coronary syndromes
▪Mural thrombi in a coronary
artery can also embolize
Coronary thrombosis