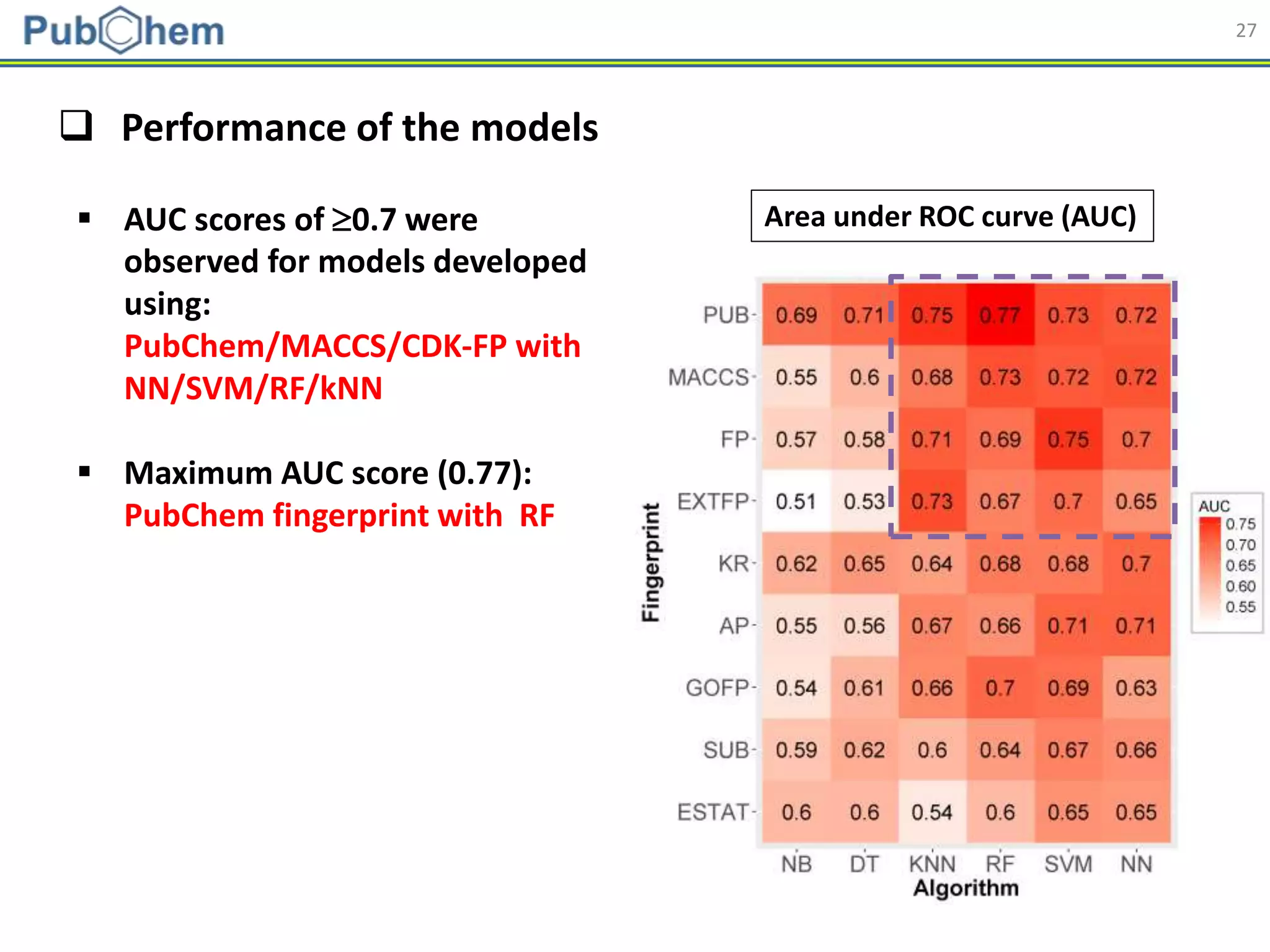
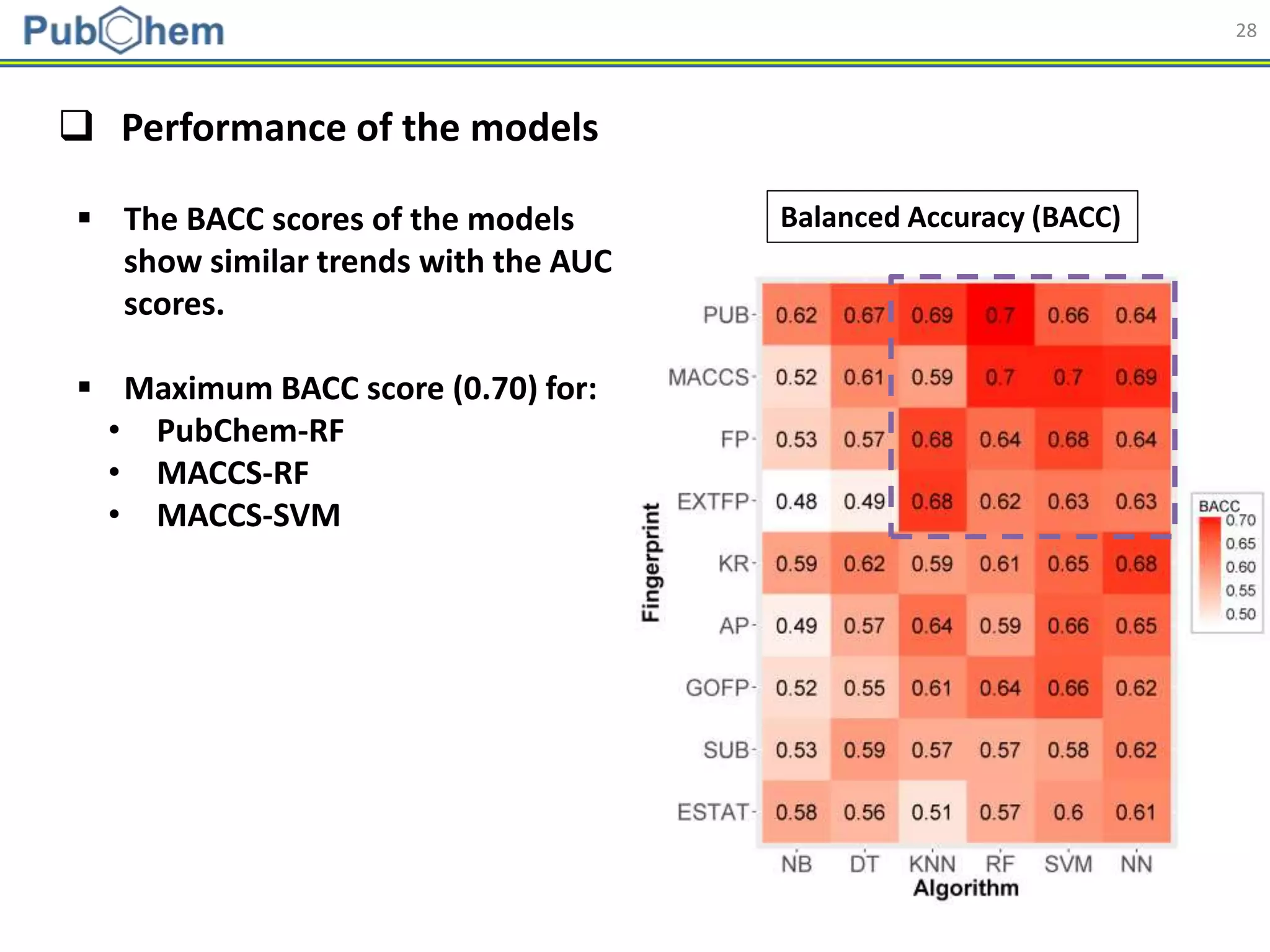
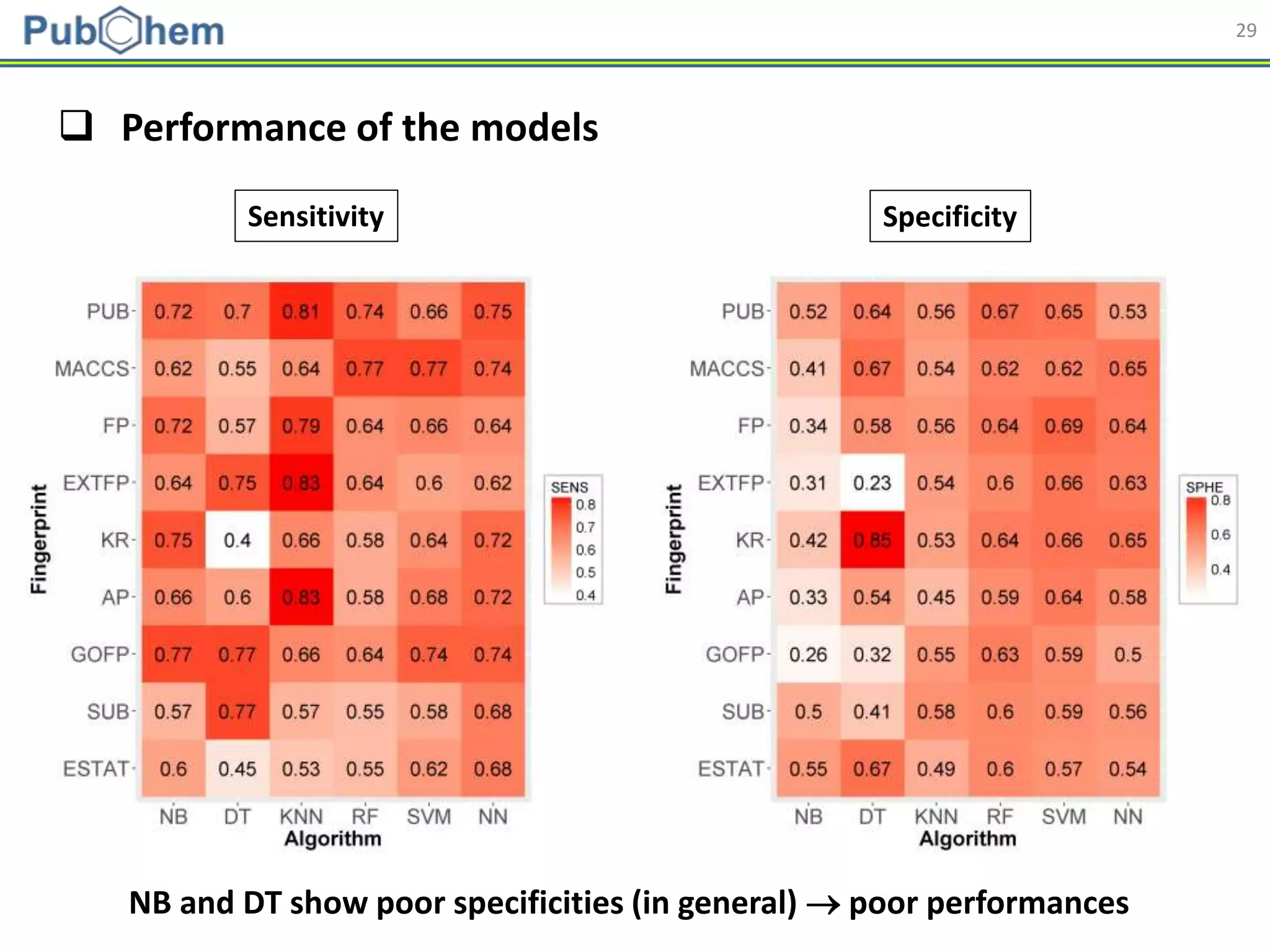
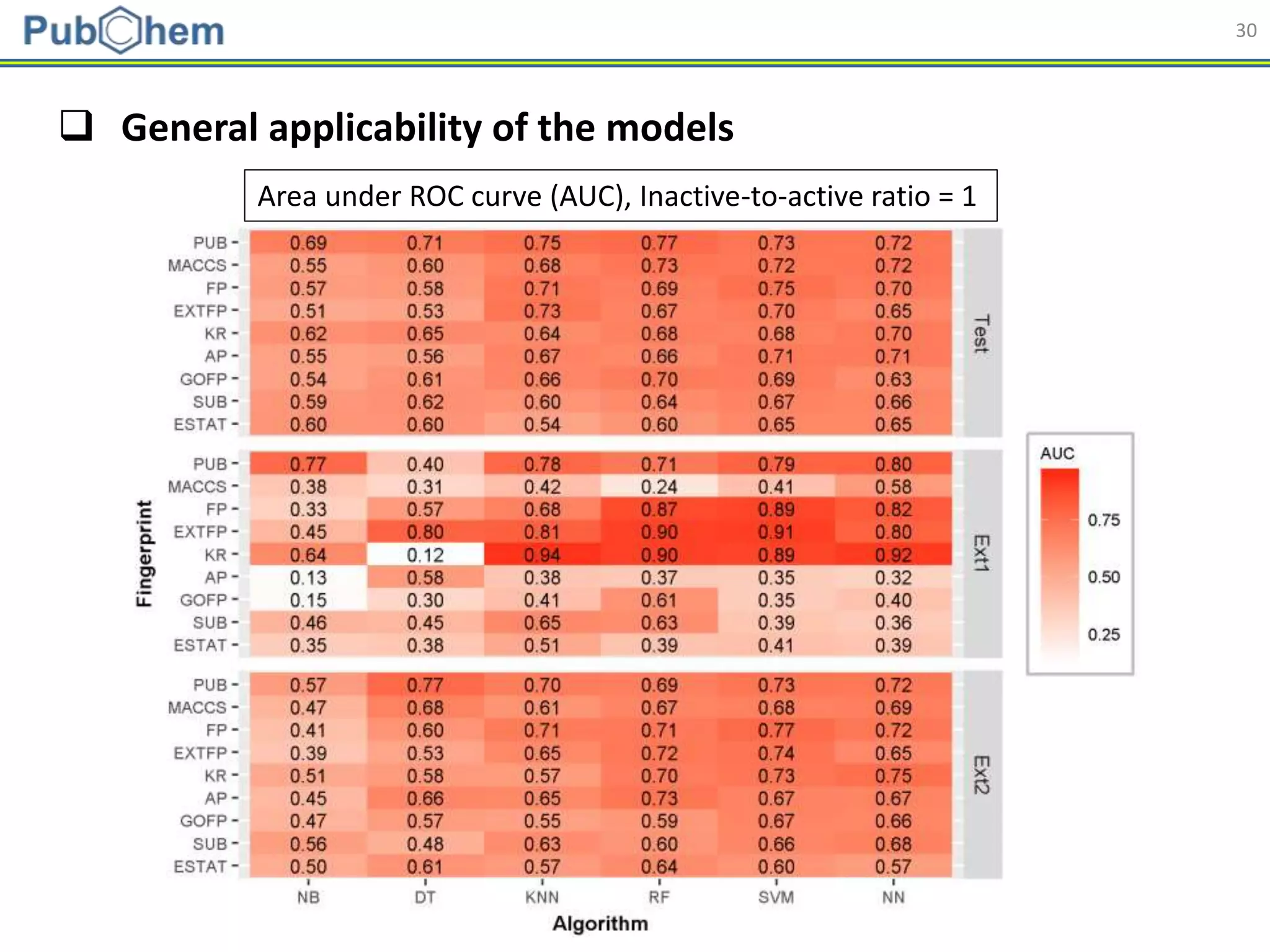
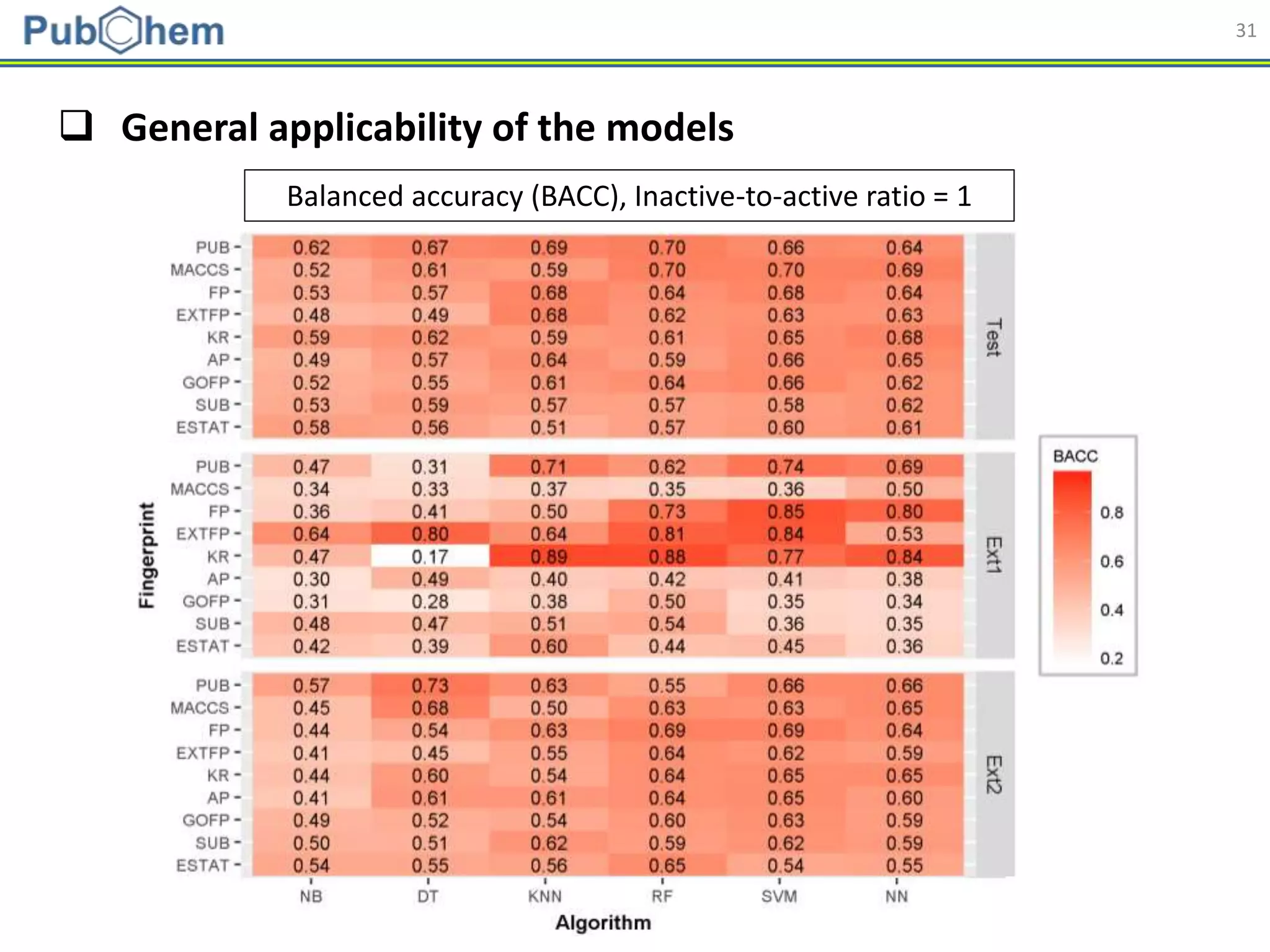
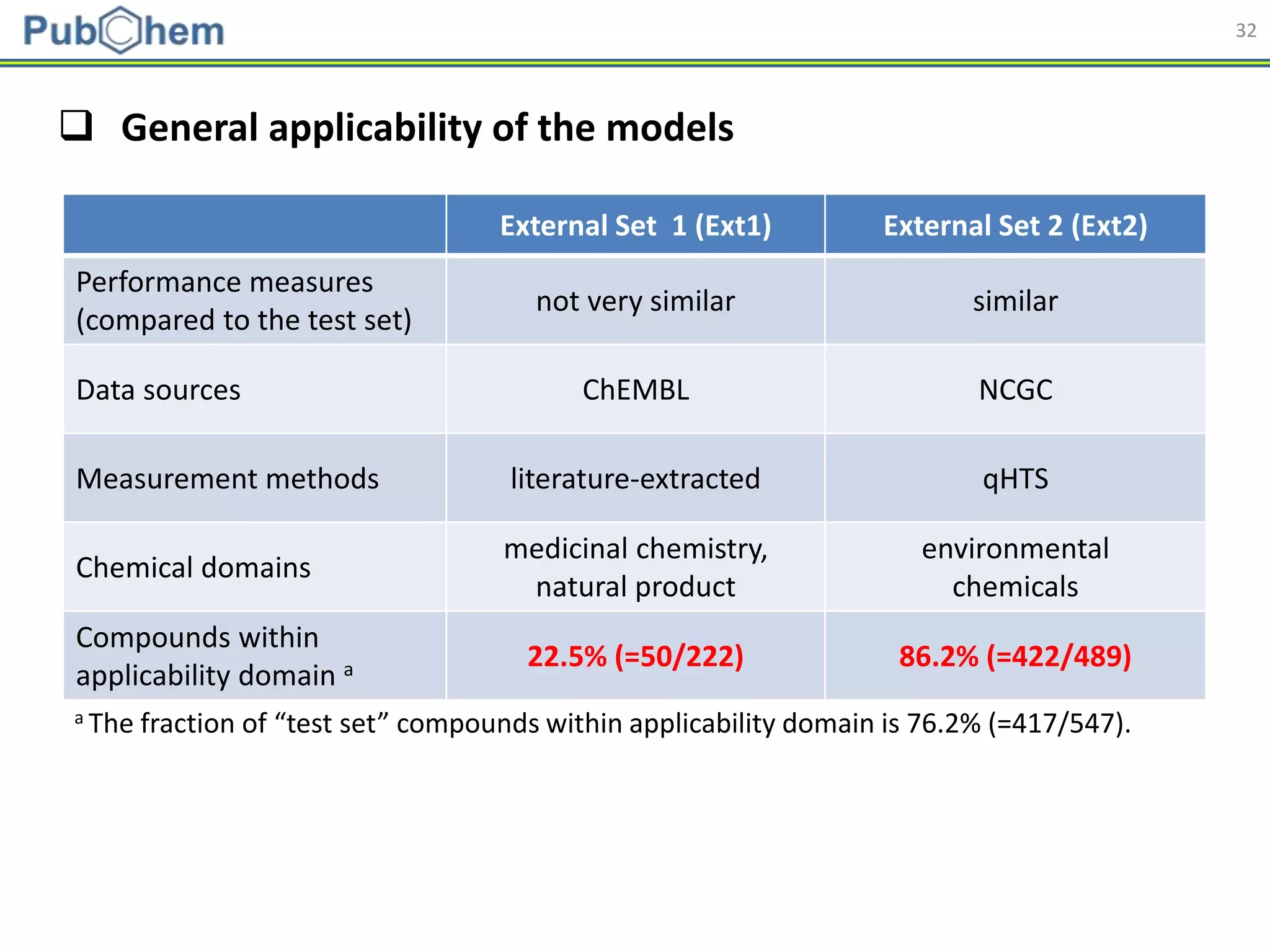
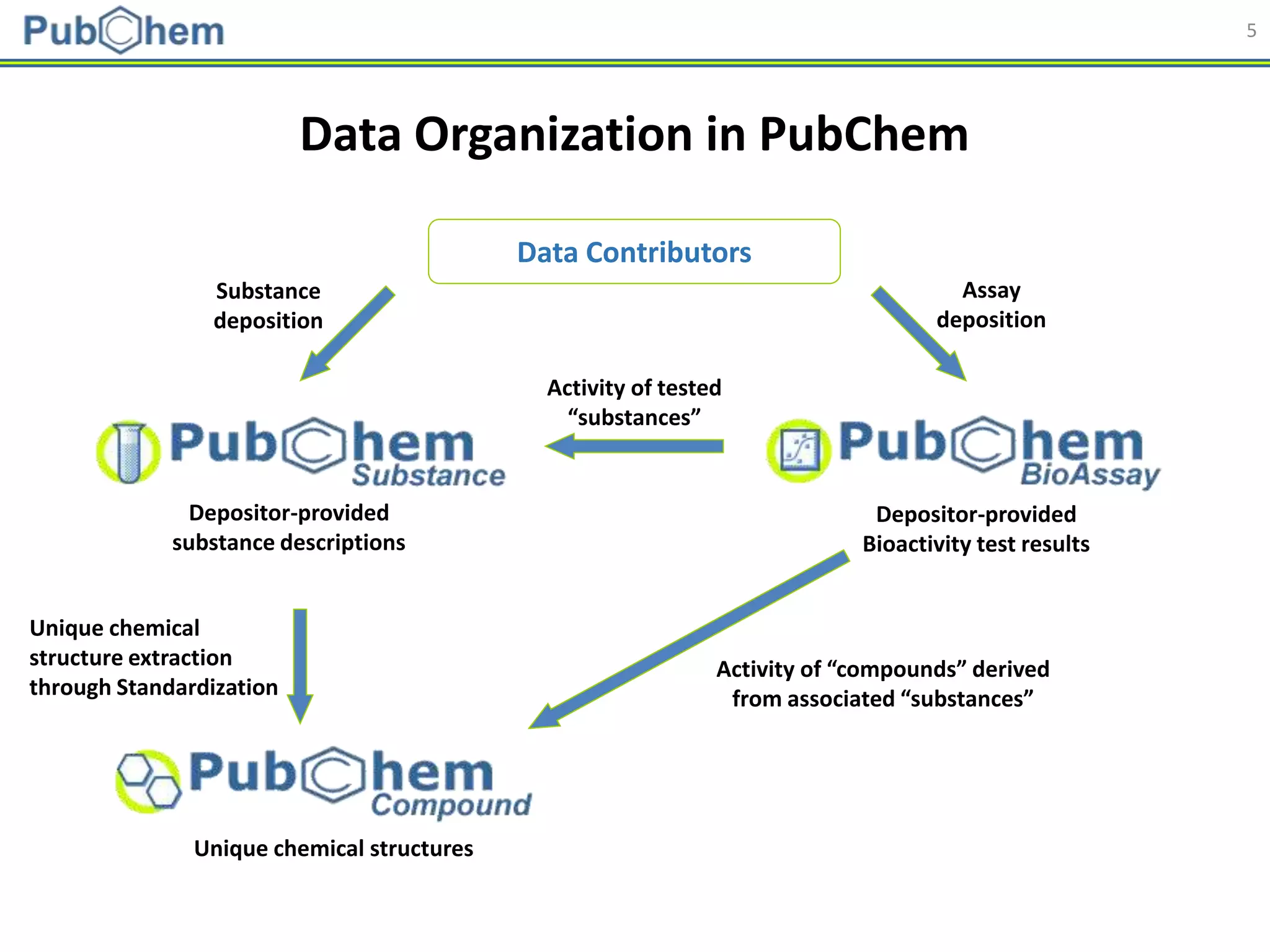
This document discusses the development of machine-learning prediction models for chemical modulators of the retinoid x receptor (RXR) signaling pathway using bioactivity data from PubChem. It outlines the methodology, including data preprocessing and model evaluation, leading to the conclusion that the best performing model achieved an AUC score of 0.77 using the random forest algorithm and PubChem fingerprint. The study highlights the potential of utilizing open bioactivity data for predictive modeling in chemical biology.














![21
Molecular descriptors
• Generated using PaDEL
[Yap CW (2011). J. Comput. Chem., 32 (7): 1466-1474]
Model Building
Abbreviation Name Length
AP AtomPairs 2D Fingerprint 780
ESTAT Estate fingerprint 79
EXTFP* CDK Extended Fingerprint 1,024
FP* CDK fingerprint 1,024
GOFP* CDK graph only fingerprint 1,024
KR Klekota-Roth fingerprint 4,860
MACCS MACCS fingerprint 166
PUB PubChem fingerprint 881
SUB Substructure fingerprint 307
* Hashed fingerprints](https://image.slidesharecdn.com/acs2018fallbostonrxrafinal-180823022938/75/Using-open-bioactivity-data-for-developing-machine-learning-prediction-models-for-chemical-modulators-of-the-retinoid-X-receptor-RXR-signaling-pathway-21-2048.jpg)



![25
Applicability Domain
𝐷 𝑇 = 𝑑 + 𝑍 ∙ 𝜎
Applicability domain of the developed models was assessed
using a distance-based approach [Shen et al. (2002). J. Med.
Chem., 45(13):2811-2823].
1. For each training set, the (Manhattan) distance to its nearest neighbor
was collected and used to determine the applicability domain threshold.
2. If a test/external set compound does not have a neighbor in the training
set (closer than DT), it was considered to be out of the applicability
domain of the model, and the prediction for that test compound was
deemed to be unreliable.](https://image.slidesharecdn.com/acs2018fallbostonrxrafinal-180823022938/75/Using-open-bioactivity-data-for-developing-machine-learning-prediction-models-for-chemical-modulators-of-the-retinoid-X-receptor-RXR-signaling-pathway-25-2048.jpg)