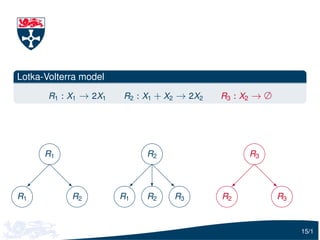
The document describes Gillespie's algorithm for simulating stochastic kinetic models. It discusses how the algorithm works and that as the number of reactions and species increases, the time per iteration increases. It proposes representing the dependencies between reactions using a directed graph to only update hazard functions that depend on changed species after a reaction occurs, improving efficiency. This is illustrated using examples of simple models.










![The direct method
1. Initialisation: initial conditions, reactions constants, and random
number generators
2. Propensities update: Update each of the M hazard functions, hi (x )
3. Propensities total: Calculate the total hazard h0 = ∑M 1 hi (x )
i=
4. Reaction time: τ = −ln [U (0, 1)]/h0 and t = t + τ
5. Reaction selection: A reaction is chosen proportional to it’s hazard
6. Reaction execution: Update species
7. Iteration: If the simulation time is exceeded stop, otherwise go back
to step 2
Typically there are a large number of iterates.
10/1](https://image.slidesharecdn.com/talk-120608045102-phpapp01/85/Speeding-up-the-Gillespie-algorithm-11-320.jpg)
















![Step 4: Reaction time
Reaction time: τ = −ln [U (0, 1)]/h0 . As the number of reactions and
species increase, the time of this step is constant.
For the toy model, we spend about 3% of computer time executing
this step
You could generate the random numbers on a separate thread (on a
multicore machines) to save you a small amount of time
20/1](https://image.slidesharecdn.com/talk-120608045102-phpapp01/85/Speeding-up-the-Gillespie-algorithm-29-320.jpg)


![Step 5: Reaction selection
Consider the following pieces of R code:
}
## u are U(0, 1) RNs
for (i in 1: length (u)) {
i f (u[i] 0.01)
x = 1
else i f (u[i]0.05)
x = 2
else i f (u[i]0.1)
x = 3
else
x = 4
Calling this piece of code 107 times takes about 34 seconds.
22/1](https://image.slidesharecdn.com/talk-120608045102-phpapp01/85/Speeding-up-the-Gillespie-algorithm-32-320.jpg)
![Step 5: Reaction selection
Now lets just reverse the order of the if statements
}
for (i in 1: length (u)) {
i f (u[i] 0.9)
x = 1
else i f (u[i]0.95)
x = 2
else i f (u[i]0.99)
x = 3
else
x = 4
Calling this piece of code 107 times takes about 15 seconds. A reduction
of around 44%.
23/1](https://image.slidesharecdn.com/talk-120608045102-phpapp01/85/Speeding-up-the-Gillespie-algorithm-33-320.jpg)














































































