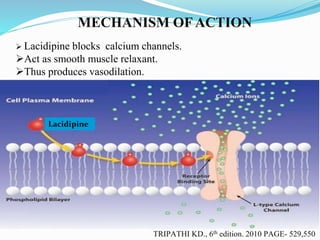
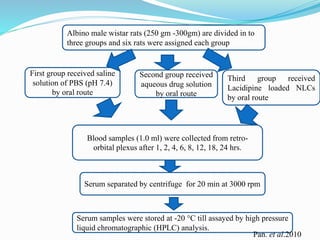
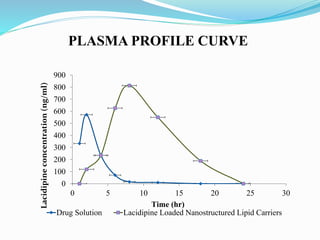
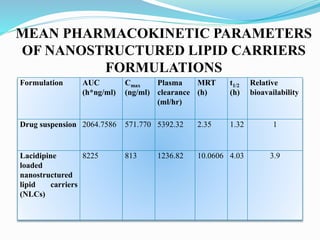
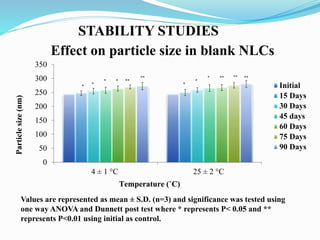
The document outlines a plan to develop nanostructured lipid carriers (NLCs) loaded with the antihypertensive drug lacidipine to enhance its oral bioavailability. Key aspects of the plan include:
1) Conducting preformulation studies such as determining lacidipine's solubility, partition coefficient, and developing HPLC and UV methods for analysis.
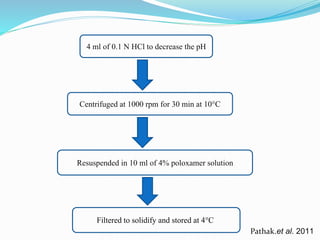
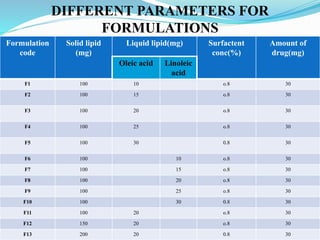
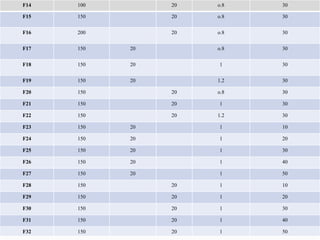
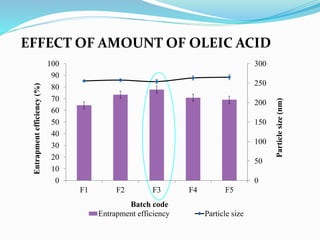
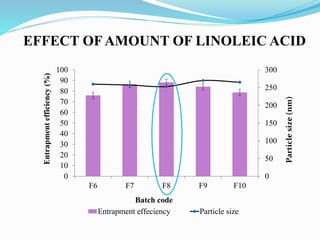
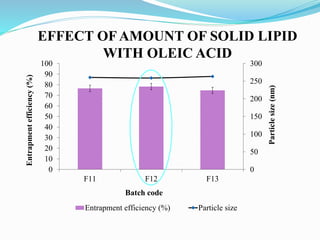
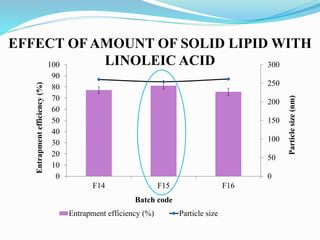
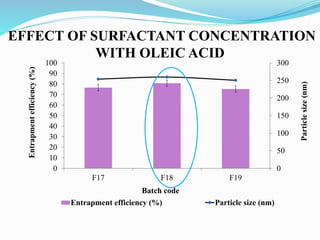
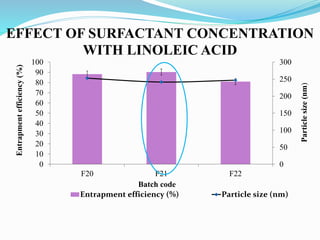
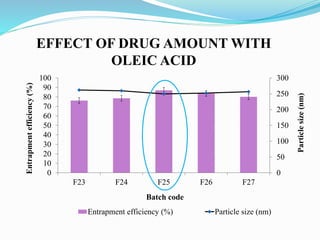
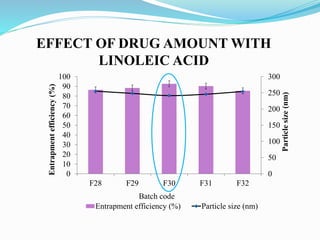
2) Preparing NLCs using a solvent injection method and studying the effect of formulation variables like lipid type and amount, surfactant concentration, and drug loading.
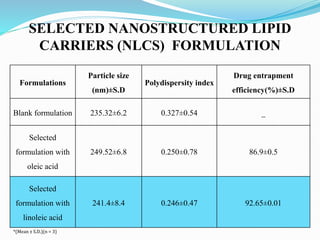
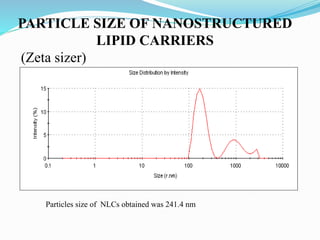
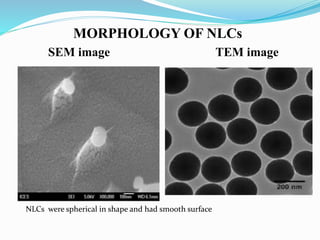
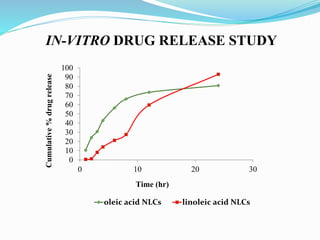
3) Characterizing the optimized NLCs using techniques like particle size analysis, drug entrapment efficiency tests, and in vitro drug release studies.











![B.P 2007, page-1205
IUPAC Name Diethyl 4-{2-[(tert butoxycarbonyl)vinyl]phenyl}- 1,4
dihydropyridine -3,5dicarboxylate
Category Antihypertensive(Calcium channel blocker)
Appearance It's a white to pale yellow colored crystalline powder
Molecular Formula C26H33NO6
Molecular Weight 455.54 g/mol
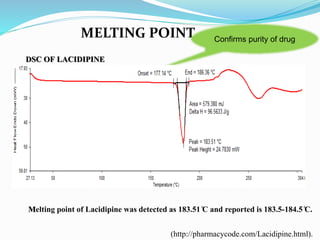
Melting point 183.5-184.5°C
Partition Coefficient 5.49
Dose 2-6mg
Bioavailabity 10%
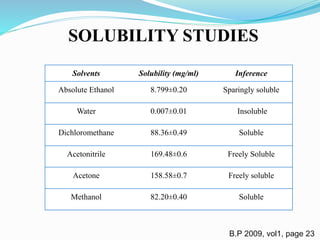
Solubility Practically insoluble in water, springly soluble in alcohol,
soluble in dichloromethane, freely soluble in acetonitrile
and acetone.
LACIDIPINE PROFILE](https://image.slidesharecdn.com/anufinal-230921093541-346df899/85/SLN-pptx-13-320.jpg)