Downloaded 48 times




























![Stabilization
34 RNeasy Mini Handbook 06/2012
Protocol: Stabilization of RNA in Harvested Animal
Tissues
This protocol describes how to stabilize and store human and animal tissues in RNAlater
RNA Stabilization Reagent, included in the RNeasy Protect Mini Kit. For RNA
purification from stabilized tissues, see “Protocol: Purification of Total RNA from Animal
Tissues”, page 37.
Important notes about RNAlater RNA Stabilization Reagent
RNA in harvested animal tissue is not protected until the tissue is completely submerged
in a sufficient volume of RNAlater RNA Stabilization Reagent. After harvesting, the
tissue should be immediately placed in at least 10 volumes of the reagent (or
approximately 10 µl reagent per 1 mg tissue). Larger volumes can be used if necessary
or desired. Smaller volumes may lead to RNA degradation during storage. Storage
containers should be wide enough so that the reagent covers the entire tissue. Storage
containers or tubes with large diameters may require more reagent to completely cover
the tissue. The procedures for tissue harvesting and RNA stabilization should be carried
out as quickly as possible.
Tissue size is critical for successful RNA stabilization with RNAlater RNA Stabilization
Reagent. Immediately upon contact, the reagent diffuses into the surface layer and outer
portions of solid tissues. To ensure rapid and reliable stabilization of RNA even in the
inner parts of solid tissues, the sample must be cut into slices less than 0.5 cm thick. The
slices can be any convenient size, provided one dimension of the sample is <0.5 cm.
If the slices are thicker than 0.5 cm, the reagent will diffuse too slowly into the interior
of the sample and RNA degradation will occur. Small organs such as rat kidney and
spleen or most mouse organs (except liver) do not require slicing: the entire organ can
be placed in RNAlater RNA Stabilization Reagent.
The following guide may help you to determine the amount of RNAlater RNA
Stabilization Reagent required for RNA stabilization:
A cube of rat kidney with a 5 mm edge length ([5 mm]3
= 125 mm3
= 125 µl)
weighs 150–175 mg and requires at least 1.5–1.75 ml of the reagent.
A 3 mm cube ([3 mm]3
= 27 mm3
= 27 µl) of most animal tissues weighs 30–35 mg
and requires at least 300–350 µl of the reagent.
Although weighing tissues is generally more accurate, RNA in unstabilized tissues will
degrade during weighing. In some cases, however, it may be more convenient to
quickly estimate the weight of tissue pieces. Average weights of various entire adult
mouse organs and the corresponding amounts of RNAlater RNA Stabilization Reagent
to use are given in Table 7.](https://image.slidesharecdn.com/qiagenhandbooks-140602220751-phpapp02/85/Qiagen-handbooks-34-320.jpg)









![Yeast
44 RNeasy Mini Handbook 06/2012
Do not overload the RNeasy spin column, as this will significantly reduce RNA yield and
purity.
Yeast growth is usually measured using a spectrophotometer. However, it is very difficult
to give specific and reliable recommendations for the relationship between OD values
and cell numbers in yeast cultures. Cell density is influenced by a variety of factors (e.g.,
species, media, and shaker speed), and OD readings of cultures measure light
scattering rather than absorption. Measurements of light scattering are highly
dependent on the distance between the sample and the detector and therefore readings
vary between different types of spectrophotometer. In addition, different species show
different OD values at defined wavelengths (e.g., 600 nm or 436 nm).
We therefore recommend calibrating the spectrophotometer by comparing OD
measurements at appropriate wavelengths with viable cell densities determined by
plating experiments (e.g., Ausubel, F.M. et al., eds. [1991] Current Protocols in
Molecular Biology. New York: Wiley Interscience). OD readings should be between
0.05 and 0.3 to ensure significance. Samples with readings above 0.3 should be
diluted so that the readings fall within this range; the dilution factor should then be used
to calculate the number of cells per milliliter.
The following values may be used as a rough guide. An S. cerevisiae culture containing
1–2 x 107
cells per milliliter, diluted 1 in 4, gives an OD600 value of approximately 0.25
with a Beckman DU®
-7400 spectrophotometer or 0.125 with a Beckman DU-40
spectrophotometer. These correspond to calculated OD values of 1 or 0.5, respectively,
for 1–2 x 107
yeast cells per milliliter.
Important points before starting
If using the RNeasy Kit for the first time, read “Important Notes” (page 16).
If working with RNA for the first time, read Appendix A (page 61).
Yeast cells should be harvested in log-phase growth. If performing enzymatic lysis
(step 1a or 1b), use only freshly harvested cells. If performing mechanical
disruption, cell pellets can be stored at –70°C for later use or used directly in the
procedure. Homogenized cell lysates from step 1c can be stored at –70°C for
several months. Frozen lysates should be incubated at 37°C in a water bath until
completely thawed and salts are dissolved. Avoid prolonged incubation, which
may compromise RNA integrity. Proceed to step 2.
Buffer RLT may form a precipitate upon storage. If necessary, redissolve by
warming, and then place at room temperature (15–25°C).
Buffer RLT and Buffer RW1 contain a guanidine salt and are therefore not
compatible with disinfecting reagents containing bleach. See page 6 for safety
information.
After enzymatic lysis or mechanical disruption, perform all steps of the procedure
at room temperature. During the procedure, work quickly.](https://image.slidesharecdn.com/qiagenhandbooks-140602220751-phpapp02/85/Qiagen-handbooks-44-320.jpg)




















![Alternatively, gene expression analysis can be performed using QuantiFast®
Probe
Assays and the QuantiFast Probe RT-PCR Plus Kit, which includes an integrated genomic
DNA removal step.
The protocol for purification of cytoplasmic RNA from animal cells (available at
www.qiagen.com/literature/protocols/RNeasyMini.aspx) is particularly advantageous
in applications where the absence of DNA contamination is critical, since intact nuclei
are removed. Using this protocol, DNase digestion is generally not required: most of
the DNA is removed with the nuclei, and RNeasy technology efficiently removes nearly
all of the remaining small amounts of DNA without DNase treatment. However, even
further DNA removal may be desirable for certain RNA applications that are sensitive
to very small amounts of DNA (e.g., TaqMan RT-PCR analysis with a low-abundance
target). Using the cytoplasmic RNA protocol with optional DNase digestion results in
undetectable levels of DNA, even in sensitive quantitative RT-PCR analyses.
Integrity of RNA
The integrity and size distribution of total RNA purified with RNeasy Kits can be checked
by denaturing agarose gel electrophoresis and ethidium bromide* staining or by using
an Agilent 2100 bioanalyzer. The respective ribosomal RNAs should appear as sharp
bands or peaks. The apparent ratio of 28S rRNA to 18S RNA should be approximately
2:1. If the ribosomal bands or peaks of a specific sample are not sharp, but appear as
a smear towards smaller sized RNAs, it is likely that the sample suffered major
degradation either before or during RNA purification.
Appendix C: Formaldehyde Agarose Gel
Electrophoresis
The following protocol for formaldehyde agarose (FA) gel electrophoresis is routinely
used at QIAGEN and gives enhanced sensitivity for gel and subsequent analysis (e.g.,
northern blotting). A key feature is the concentrated RNA loading buffer that allows a
larger volume of RNA sample to be loaded onto the gel than conventional protocols
(e.g., Sambrook, J. et al. [1989] Molecular cloning — a laboratory manual. 2nd ed.
Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press).
* When working with chemicals, always wear a suitable lab coat, disposable gloves, and protective goggles.
For more information, consult the appropriate safety data sheets (SDSs), available from the product supplier.
RNeasy Mini Handbook 06/2012 65](https://image.slidesharecdn.com/qiagenhandbooks-140602220751-phpapp02/85/Qiagen-handbooks-65-320.jpg)
![FA gel preparation
To prepare FA gel (1.2% agarose) of size 10 x 14 x 0.7 cm, mix:
1.2 g agarose*
10 ml 10x FA gel buffer (see composition below)
Add RNase-free water to 100 ml
If smaller or larger gels are needed, adjust the quantities of components proportionately.
Heat the mixture to melt agarose. Cool to 65°C in a water bath. Add 1.8 ml of 37%
(12.3 M) formaldehyde* and 1 µl of a 10 mg/ml ethidium bromide* stock solution.
Mix thoroughly and pour onto gel support. Prior to running the gel, equilibrate in 1x FA
gel running buffer (see composition below) for at least 30 min.
RNA sample preparation for FA gel electrophoresis
Add 1 volume of 5x RNA loading buffer (see composition below) to 4 volumes of RNA
sample (e.g., 10 µl of loading buffer and 40 µl of RNA) and mix.
Incubate for 3–5 min at 65°C, chill on ice, and load onto the equilibrated FA gel.
Gel running conditions
Run gel at 5–7 V/cm in 1x FA gel running buffer.
Composition of FA gel buffers
10x FA gel buffer
200 mM 3-[N-morpholino]propanesulfonic acid (MOPS) (free acid)*
50 mM sodium acetate*
10 mM EDTA*
pH to 7.0 with NaOH*
1x FA gel running buffer
100 ml 10x FA gel buffer
20 ml 37% (12.3 M) formaldehyde
880 ml RNase-free water
* When working with chemicals, always wear a suitable lab coat, disposable gloves, and protective goggles.
For more information, consult the appropriate safety data sheets (SDSs), available from the product supplier.
66 RNeasy Mini Handbook 06/2012](https://image.slidesharecdn.com/qiagenhandbooks-140602220751-phpapp02/85/Qiagen-handbooks-66-320.jpg)






























































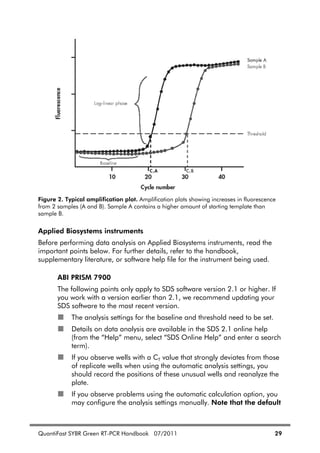
![20 QuantiFast SYBR Green RT-PCR Handbook 07/2011
Appendix B: Assay Design and Handling Primers
Important factors for successful quantitative, real-time RT-PCR include the
design of optimal primer pairs, the use of appropriate primer concentrations,
and the correct storage of primers.
Assay design
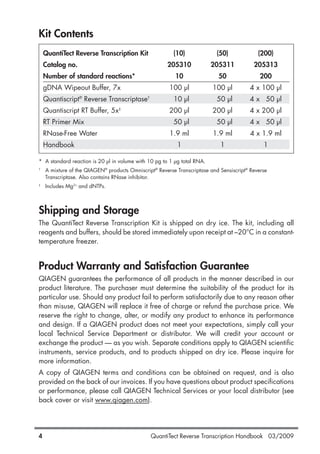
For guaranteed results in gene expression analysis experiments, we recommend
using QuantiTect Primer Assays (see Ordering Information, page 36). If
designing your own primers, please follow the guidelines provided in Table 3.
Since fluorescence from SYBR Green I increases strongly upon binding of the
dye to any double-stranded DNA, it is particularly important to minimize
nonspecific primer annealing by careful primer design.
Table 3. General Guidelines for Design of Primers
Length 18–30 nucleotides
GC content 40–60%
Tm For best results, use commercially available oligo-design
software such as OLIGO 6 (oligo.net) or Web-based tools
such as Primer3 (frodo.wi.mit.edu/cgi-bin/primer3/
primer3_www.cgi)* to determine primer Tms.
Simplified formula for estimating melting temperature (Tm):
Tm = 2ºC x (number of [A+T]) + 4ºC x (number of [G+C])
Whenever possible, design primer pairs with similar Tm
values.
Sequence Always check the specificity of primers by performing a
BLAST®
search (www.ncbi.nlm.nih.gov/blast). Ensure
that primer sequences are unique for your template
sequence.
Ensure the length of the PCR product is less than 200
bp.
Table continued on next page
* Rozen, S. and Skaletsky, H.J. (2000) Primer3 on the WWW for general users and for
biologist programmers. In: Krawetz, S. and Misener, S., eds. Bioinformatics Methods and
Protocols: Methods in Molecular Biology. Totowa, NJ: Humana Press, pp. 365–386.](https://image.slidesharecdn.com/qiagenhandbooks-140602220751-phpapp02/85/Qiagen-handbooks-132-320.jpg)


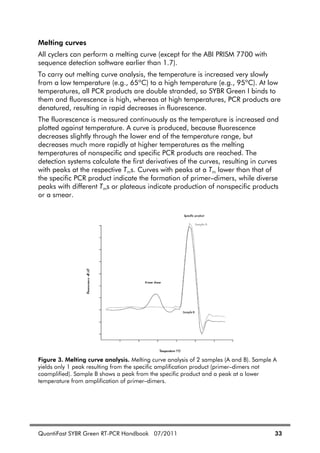
![QuantiFast SYBR Green RT-PCR Handbook 07/2011 23
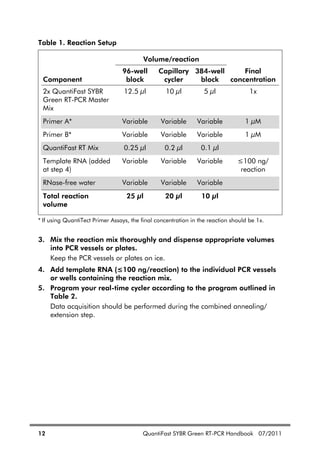
Table 4. Continued
Dissolving
primers
Before opening a tube containing lyophilized primer, spin
the tube briefly to collect all material at the bottom of the
tube. To dissolve the primer, add the required volume of
TE, mix, and leave for 20 minutes to allow the primer to
completely dissolve. Mix again and determine the
concentration by spectrophotometry as described below.
We do not recommend dissolving primers in water. They
are less stable in water and some may not dissolve easily.
Concentration Spectrophotometric conversion for primers:
1 A260 unit = 20–30 g/ml
To check primer concentration, the molar extinction
coefficient ( 260) can be used:
A260 = 260 x molar concentration of primer
If the 260 value is not given on the data sheet supplied
with the primers, it can be calculated from the primer
sequence using the following formula:
260 = 0.89 x [(A x 15,480) + (C x 7340) + (G x 11,760)
+ (T x 8850)]
Example
Concentration of diluted primer: 1 M = 1 x 10–6
M
Primer length: 24 nucleotides with 6 each of A, C, G, and
T bases
Calculation of expected A260: 0.89 x [(6 x 15,480) +
(6 x 7340) + (6 x 11,760) + (6 x 8850)] x (1 x 10–6
)
= 0.232
The measured A260 should be within +/– 30% of the
theoretical value. If the measured A260 is very different to
the theoretical value, we recommend recalculating the
concentration of the primers, or having the primers
resynthesized.
Primer quality The quality of 18–30mers can be checked on a 15%
denaturing polyacrylamide gel; a single band should be
seen. Please contact QIAGEN Technical Services or your
local distributor (see back cover) for a protocol.](https://image.slidesharecdn.com/qiagenhandbooks-140602220751-phpapp02/85/Qiagen-handbooks-135-320.jpg)


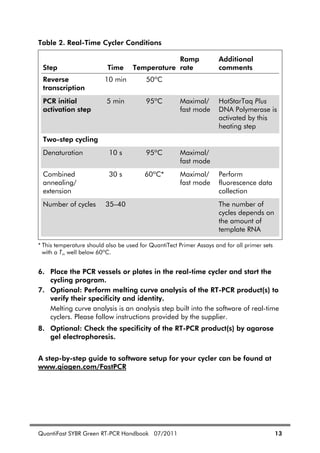
![26 QuantiFast SYBR Green RT-PCR Handbook 07/2011
Standards
For absolute quantification of RNA molecules (see page 24), the copy number
or concentration of the nucleic acids used as standards must be known. In
addition, standards should show the following features:
Primer binding sites identical to the target to be quantified
Sequence between primer binding sites identical or highly similar to target
sequence
Sequences upstream and downstream from the amplified sequence
identical or similar to “natural” target
For quantification of RNA, we strongly recommend using RNA molecules as
standards. Depending on the sequence and structure of the target and the
efficiency of reverse transcription, only a proportion of the target RNA will be
reverse transcribed. The DNA generated during reverse transcription serves as
the template for amplification in the subsequent PCR. The use of RNA standards
takes the variable efficiency of the RT reaction into account.
RNA standards can be created by cloning part or all of the transcript of interest
into a standard cloning vector. The insert can be generated by RT-PCR from
total RNA or mRNA, or by PCR from cDNA. The cloning vector must contain an
RNA polymerase promoter such as T7, SP6, or T3. Ensure that in vitro
transcription of the insert leads to generation of the sense transcript. After in
vitro transcription, plasmid DNA must be removed completely with RNase-free
DNase, since residual plasmid DNA will lead to errors in spectrophotometric
determination of RNA concentration and will also serve as a template in the
subsequent PCR. Furthermore, ensure that the RNA used as a standard does not
contain any degradation products or aberrant transcripts by checking that it
migrates as a single band in gel electrophoresis.
After determination of RNA concentration by spectrophotometry, the copy
number of standard RNA molecules can be calculated using the following
formula:
(X g/ l RNA / [transcript length in nucleotides x 340]) x 6.022 x 1023
= Y
molecules/ l
Example
Transcript length: 500 nucleotides
Concentration: 30 ng/ l = 30 x 10–9
g/ l
Calculation: (30 x 10–9
g/ l / [500 x 340]) x 6.022 x 1023
= 1.1 x 1011
molecules/ l
An alternative to the use of in vitro transcripts as RNA standards is the use of a
defined RNA preparation (e.g., from a cell line or virus preparation), for which
the absolute concentration of the target has already been determined.](https://image.slidesharecdn.com/qiagenhandbooks-140602220751-phpapp02/85/Qiagen-handbooks-138-320.jpg)

















QIAGEN provides kits for purifying RNA from a variety of sources including animal cells, tissues, bacteria, yeast, and plants. The RNeasy Mini kits use silica membrane technology to selectively bind and purify RNA over 200 nucleotides in length. Samples are lysed in a guanidine-based buffer to immediately inactivate RNases, followed by binding of RNA to the RNeasy membrane. Contaminants are washed away and high-quality RNA is eluted for downstream applications such as RT-PCR. Protocols are included for different starting materials including cells, tissues, bacteria, yeast, and plants.















































