Downloaded 1,477 times


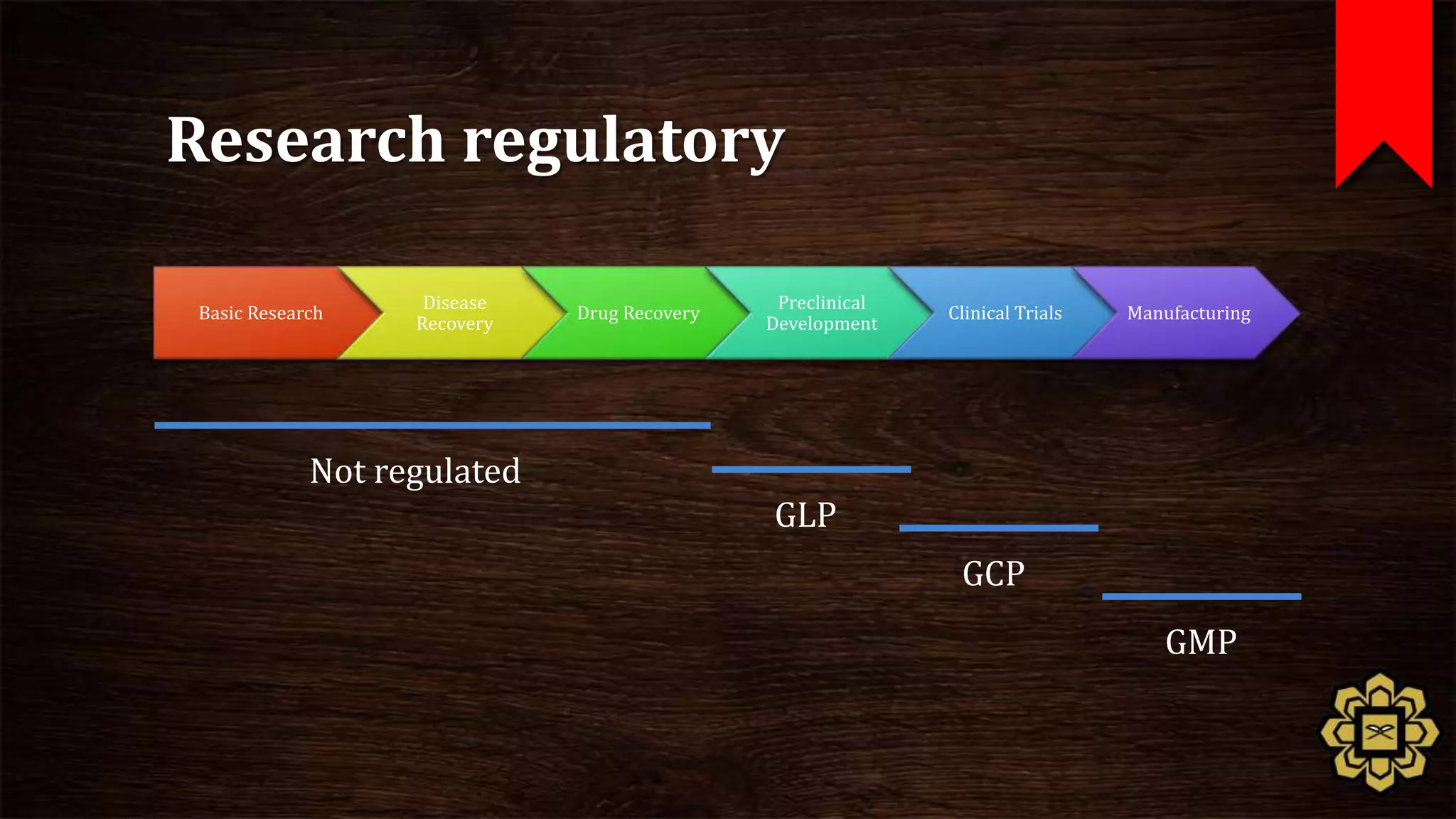
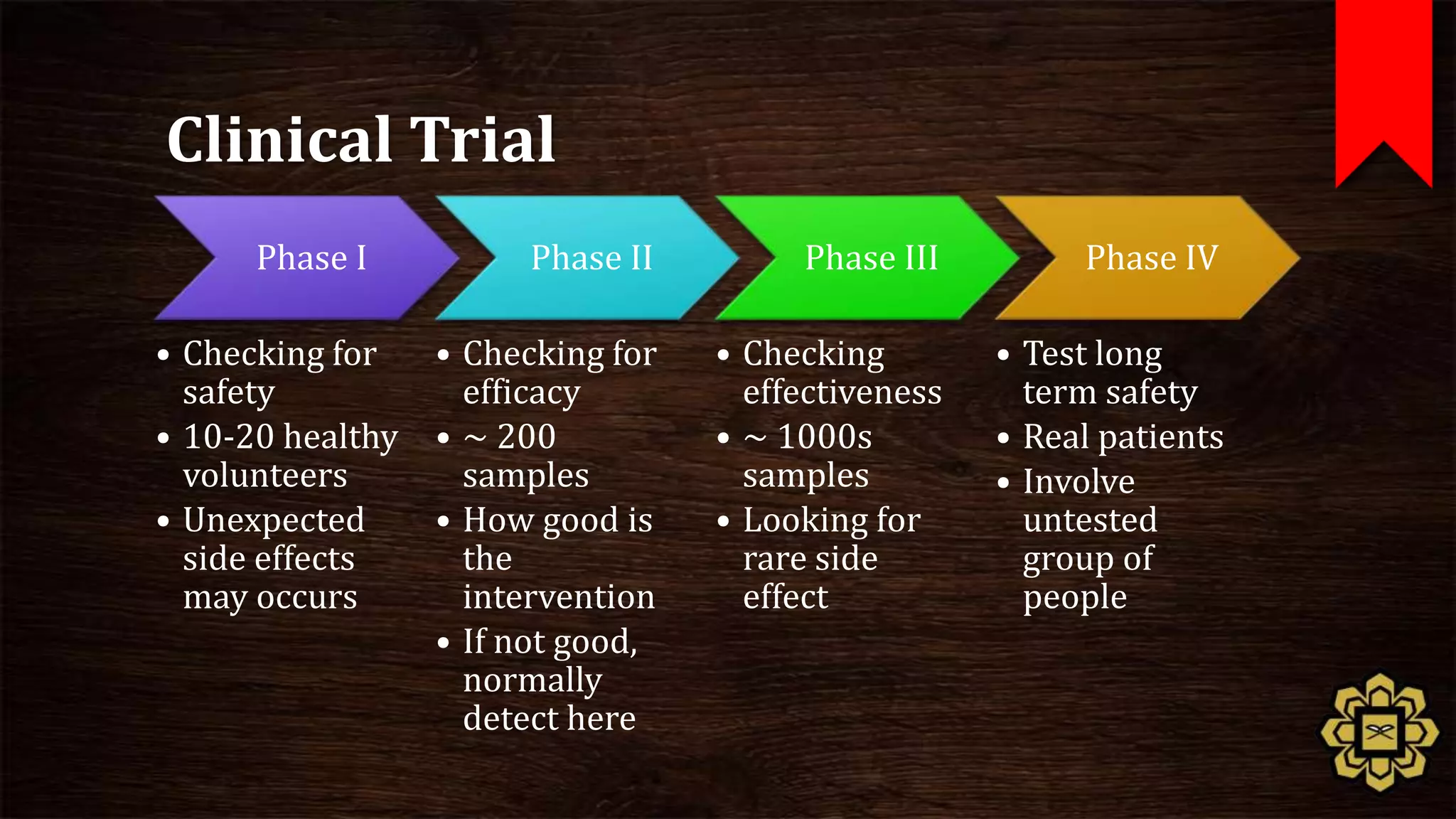












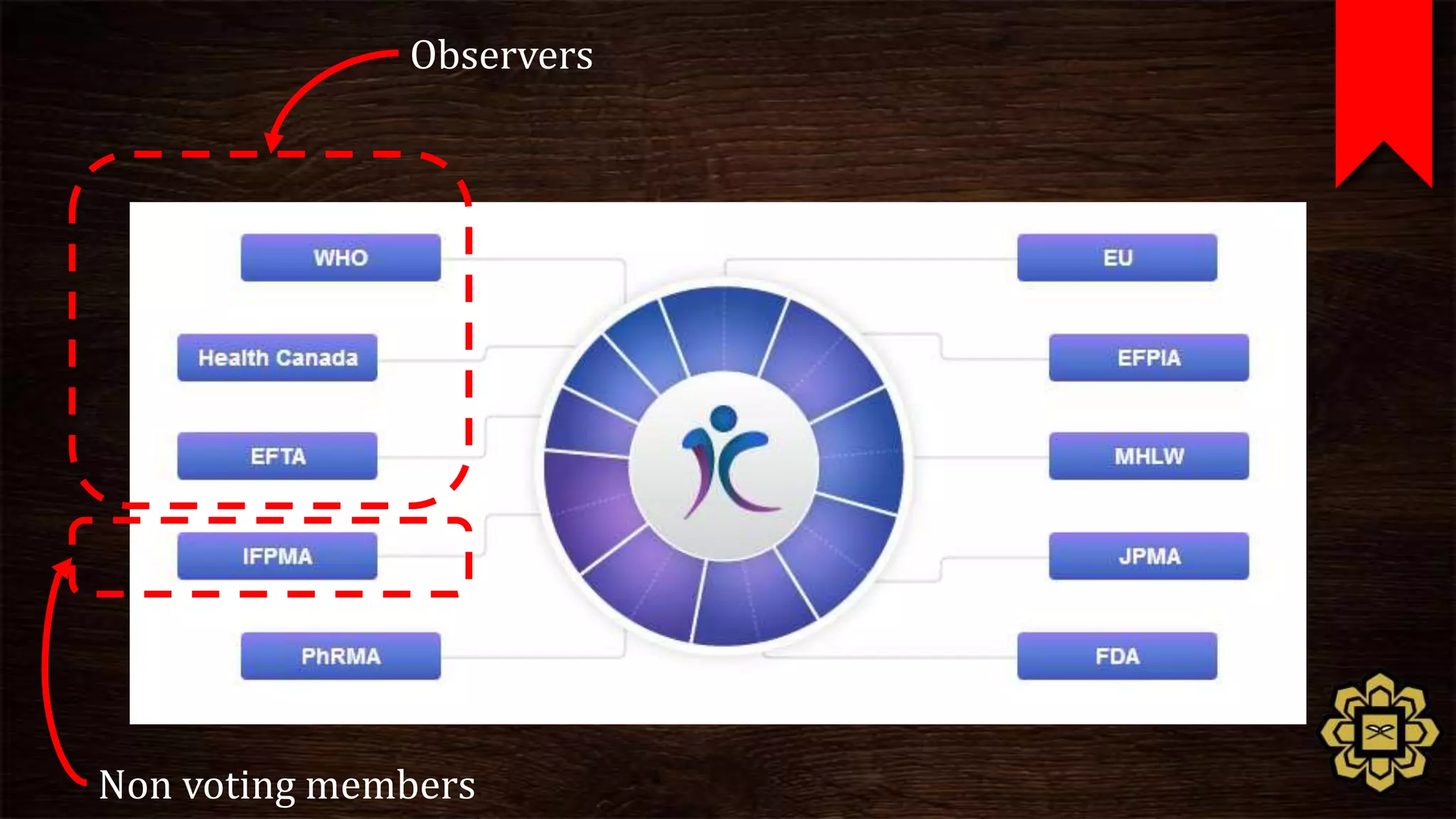














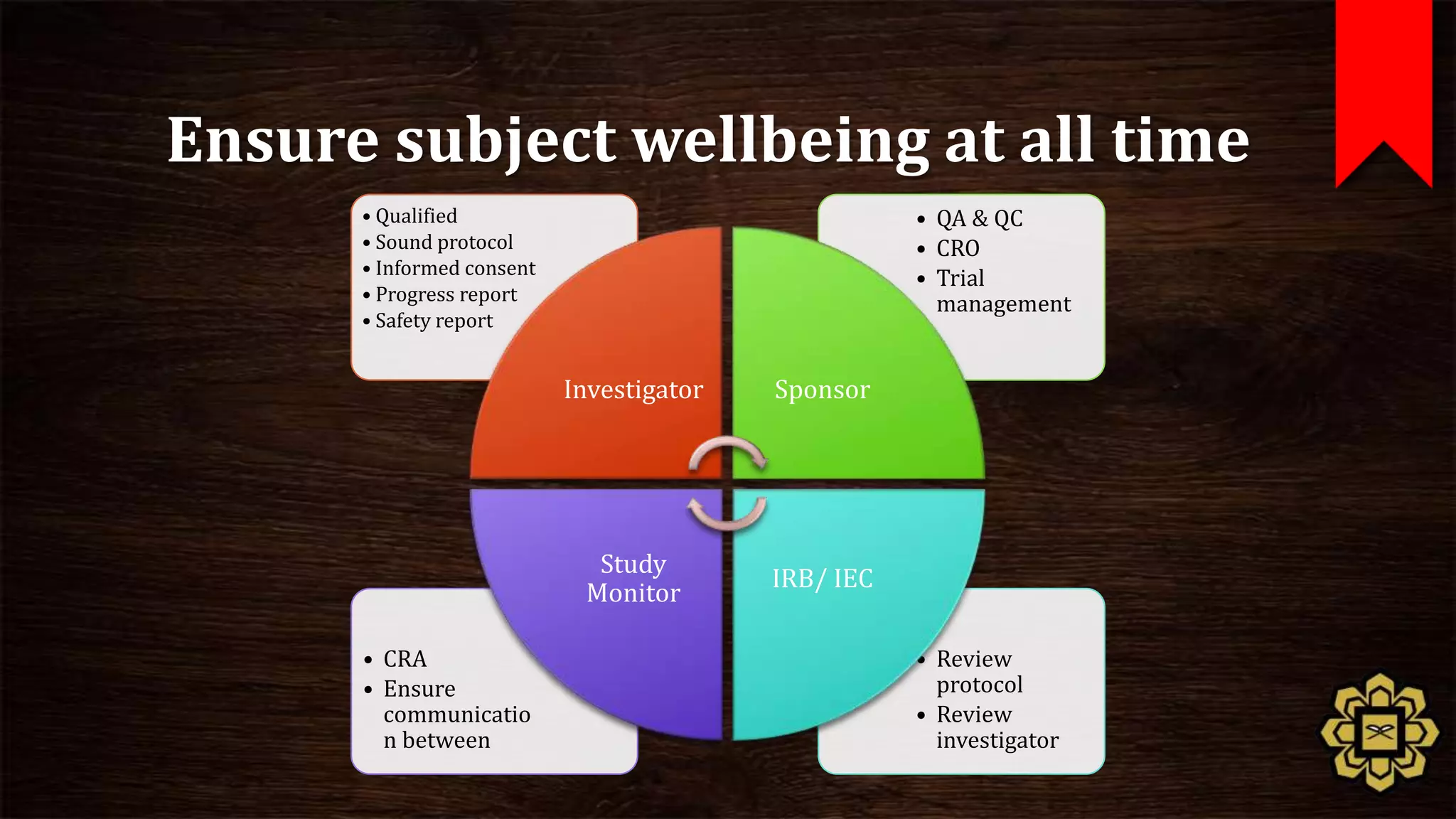


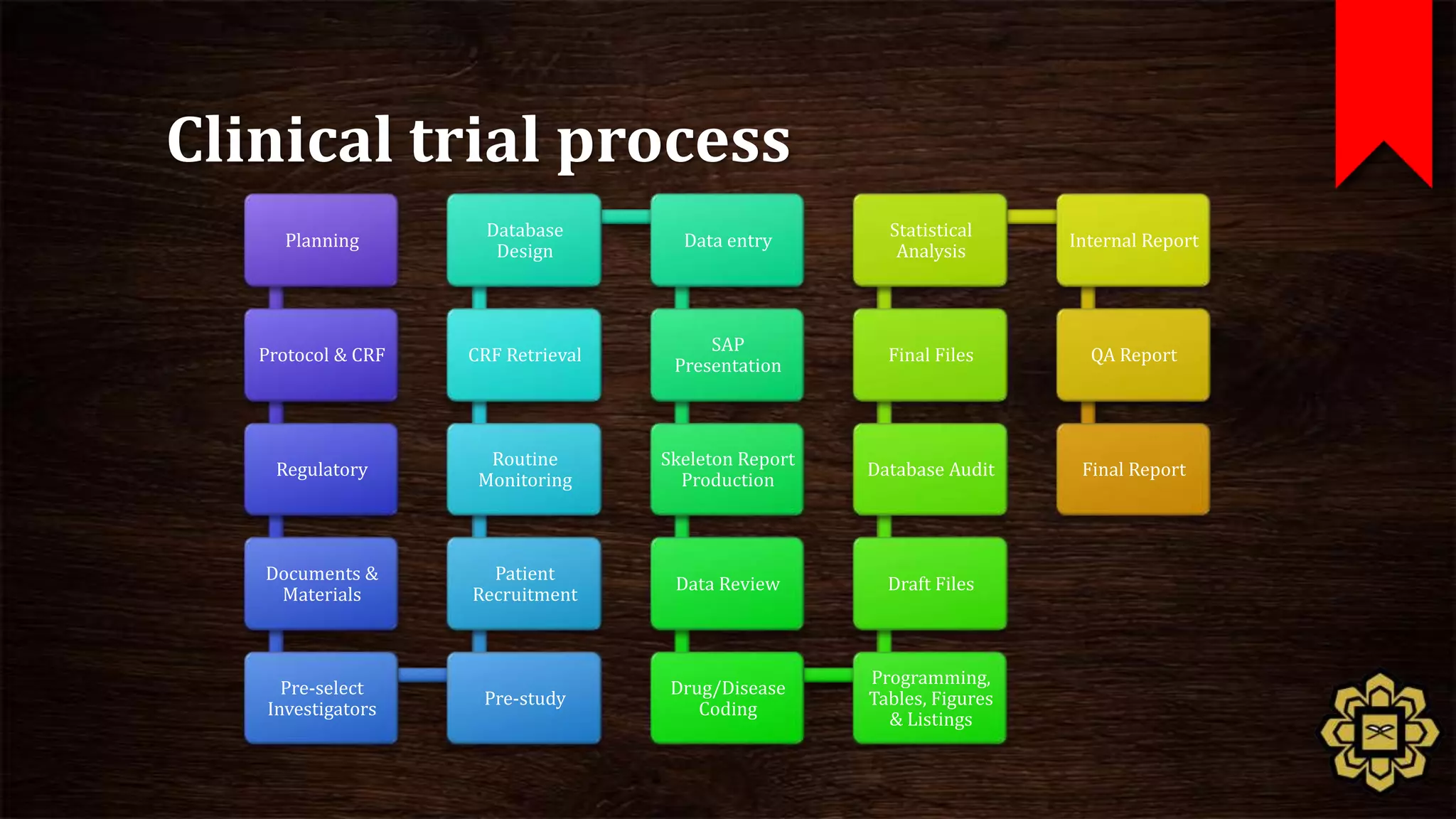





The document describes the principles of good clinical practice (GCP) according to international guidelines. It provides a brief history of human subject research regulations, from the Nuremberg Code established after World War II to modern standards like the Declaration of Helsinki, Belmont Report, and ICH GCP guidelines. The core principles of GCP outlined in the document are: prioritizing subject well-being, using qualified researchers, obtaining informed consent, maintaining quality standards, and appropriately recording and protecting private data. Adhering to GCP aims to safely and ethically conduct clinical trials involving human participants.

































































































