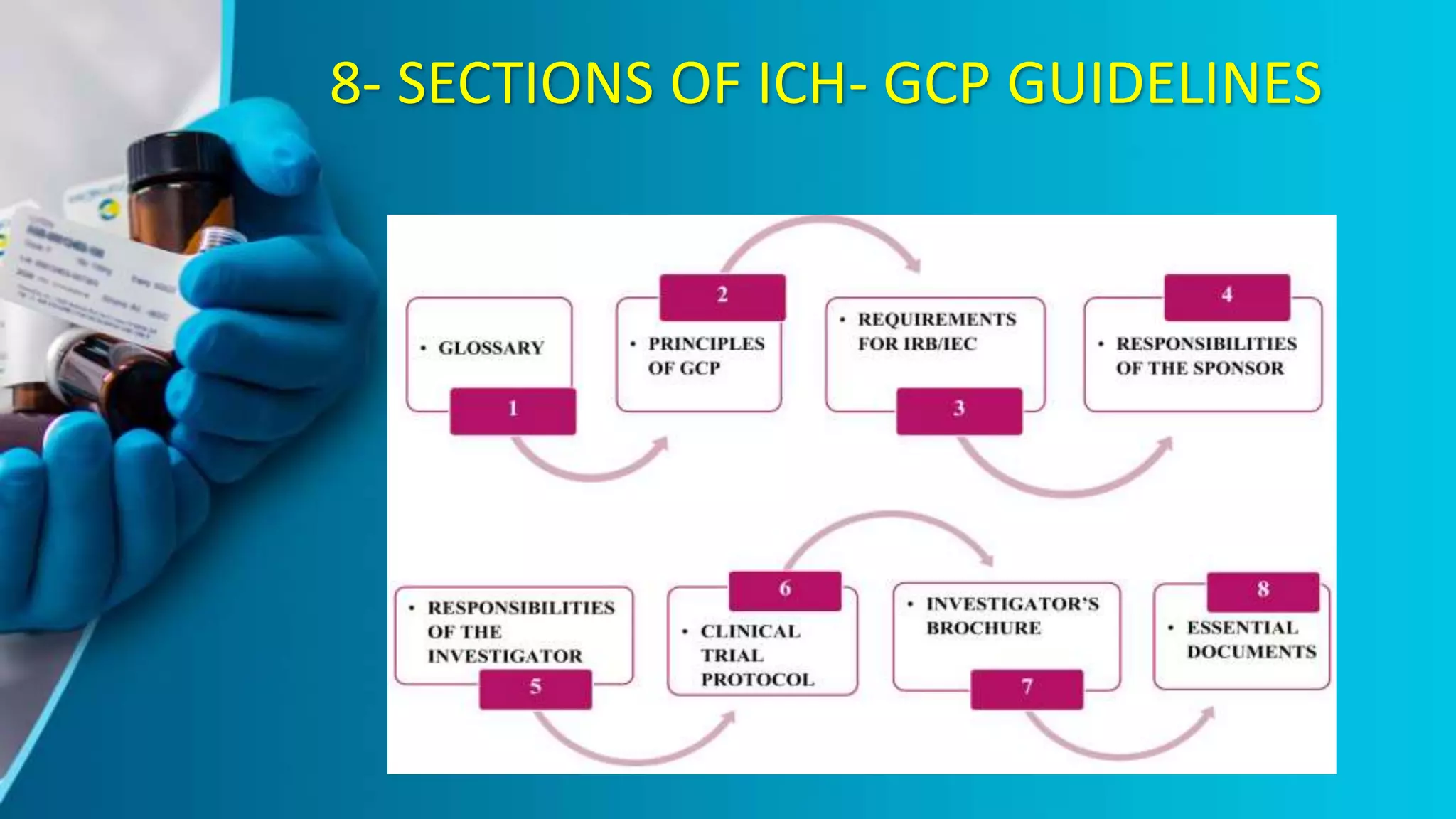
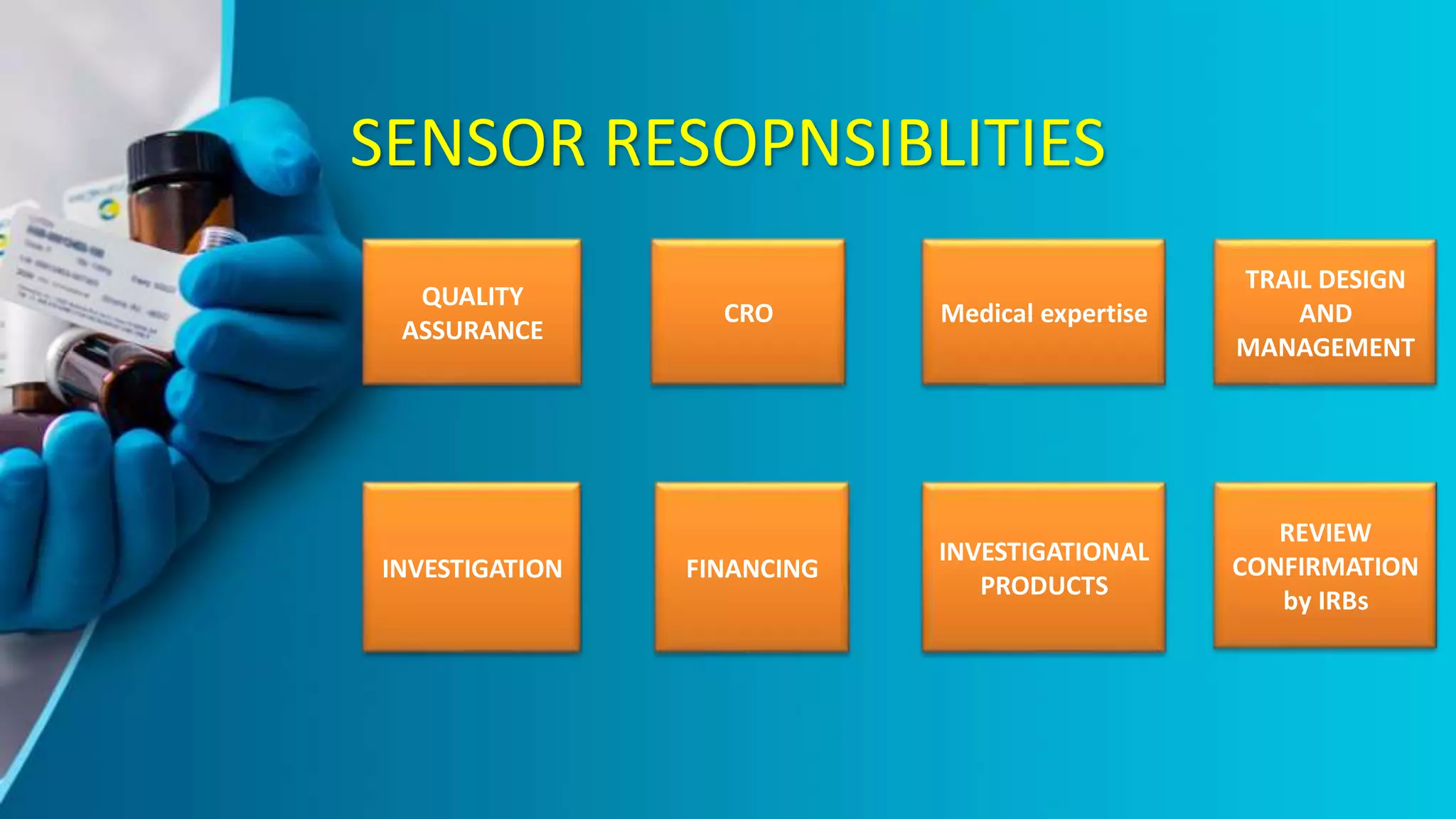
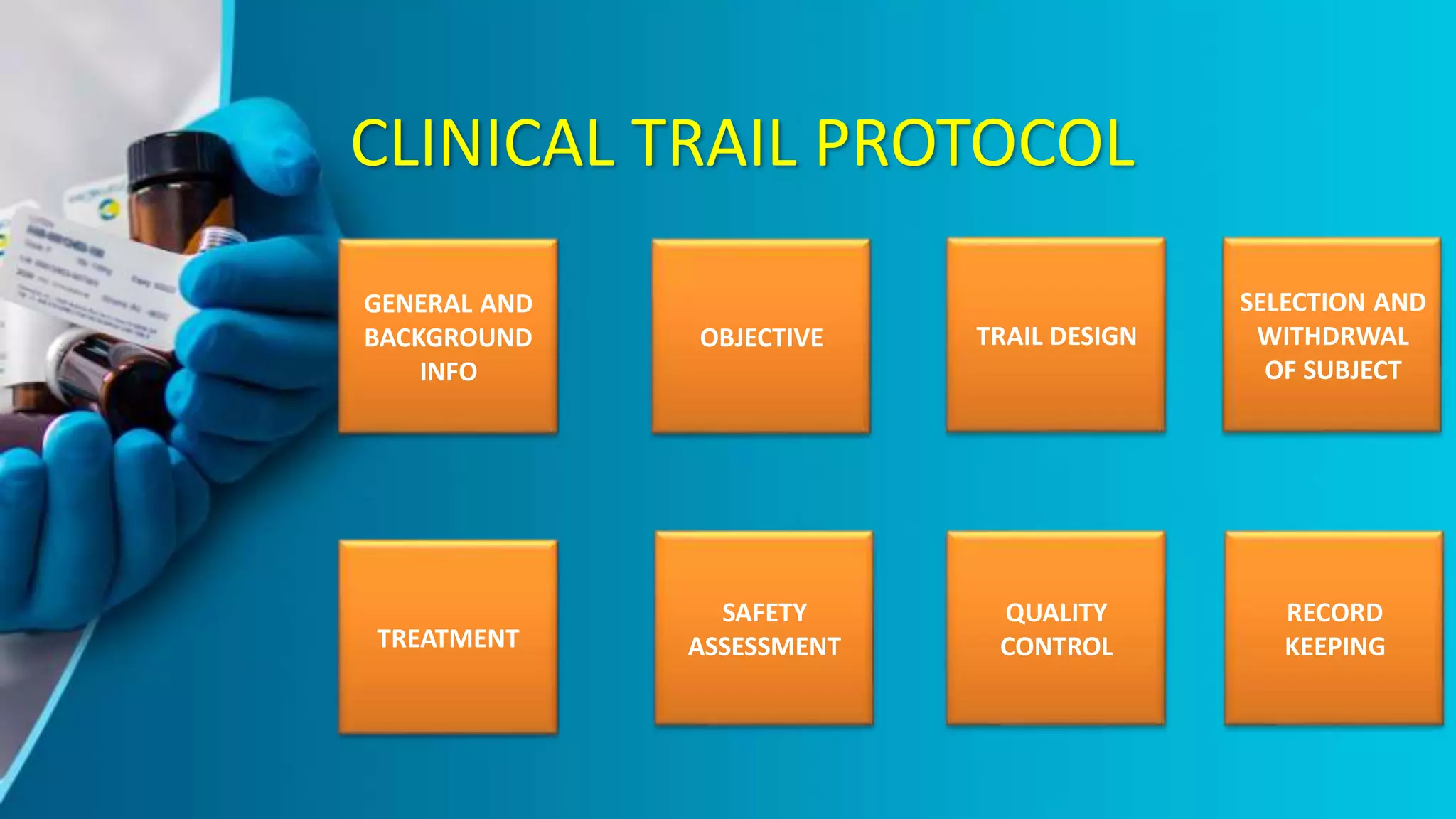
The document provides an overview of the ICH-GCP guidelines, which are international standards for conducting clinical trials to ensure the rights, safety, and well-being of participants. It outlines the principles and responsibilities pertaining to research ethics, informed consent, and the role of ethics committees, investigators, and sponsors. The guidelines aim to facilitate mutual acceptance of clinical data across major global jurisdictions, improving data quality and minimizing human exposure to risks.