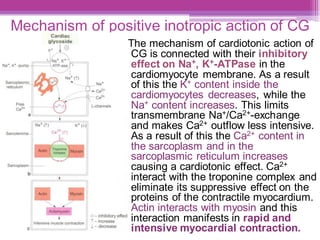
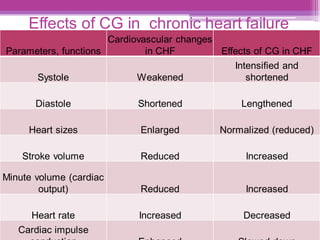
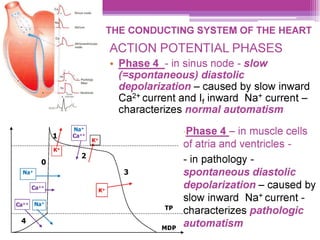
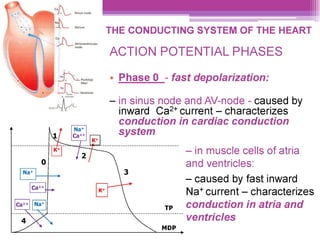
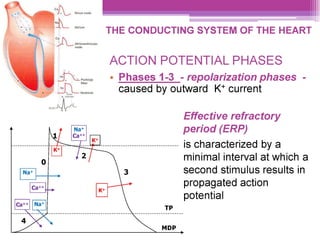
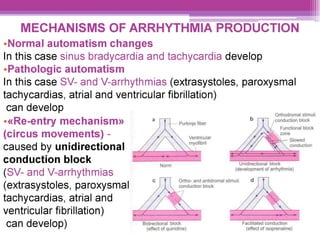
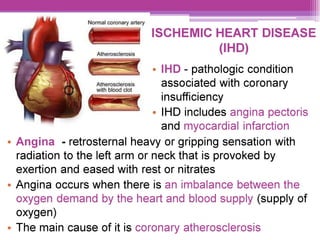
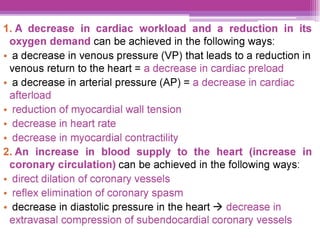
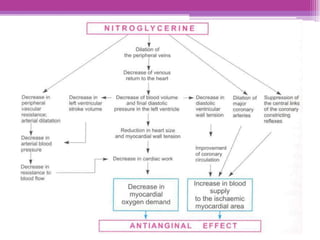
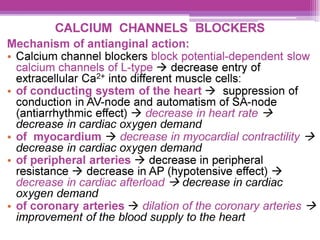
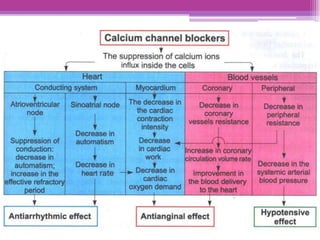
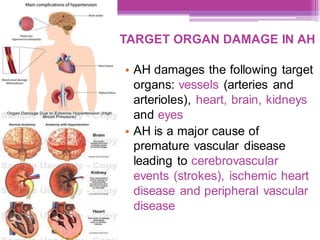
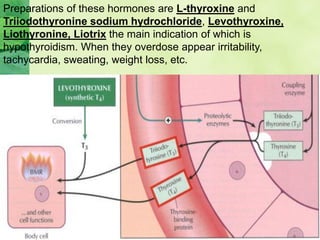
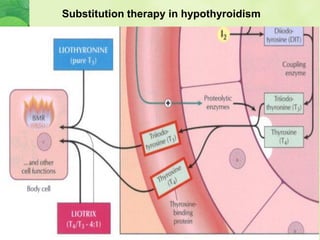
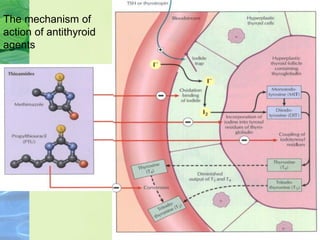
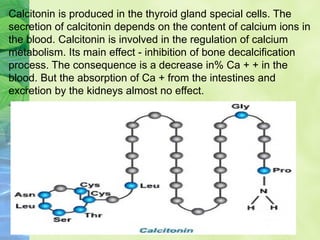
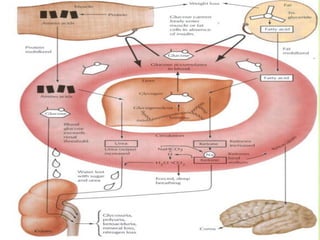
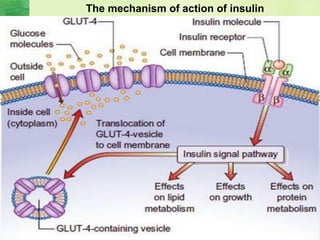
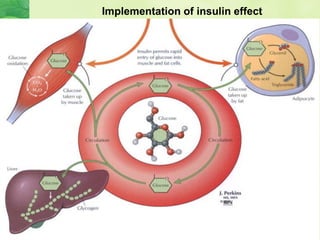
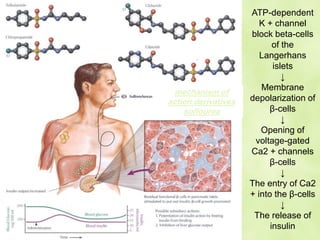
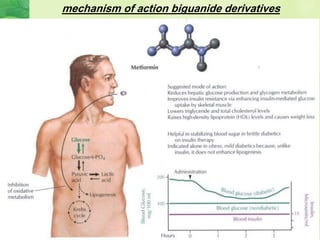
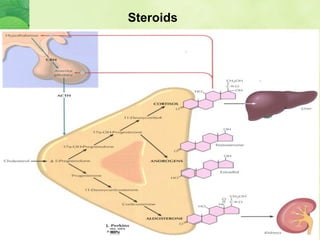
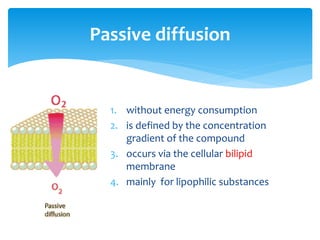
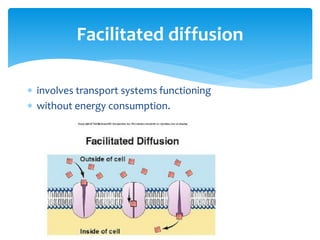
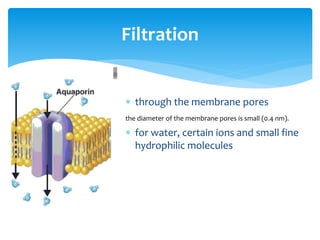
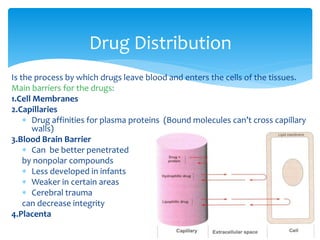
This document discusses general pharmacology and is presented by Elena Semenova, Associate Professor of Pharmacology. It addresses the key topics of pharmacokinetics, which is how the body affects drugs, and pharmacodynamics, which is how drugs affect the body. Pharmacokinetics involves absorption, distribution, metabolism and excretion of drugs in the body. Pharmacodynamics examines the biological effects of compounds and their localization and mechanisms of action. The document also outlines various drug administration routes, factors influencing drug concentrations, and the processes of drug distribution, metabolism, and elimination from the body.



































































































































































































































































![ a medicine that loosens the bowel contentsand encouragesevacuation.
1.Drugs, stimulating bowel chemoceptors:
A. Plant-based drugs
1) Drugs containing antraglycosides (CortexFrangulae, Folia Sennae, Senade)
2) Plant oils (Castor oil)
B. Synthetic drugs (Phenolphthalein, Bisacodyl, Oxyphenisatine)
2.Drugs, stimulating bowel mechanoreceptors (drugsthat are not
absorbed and increase volume of intestinalcontent):
A. Saline laxatives (Magnesium sulphate, Sodium sulphate)
B. Syntheticdrugs (polyethylene glycol [macrogol4000 (forlax)]
C. Plant-based drugs (cellulose from fruits and vegetables)
3. Faecal softeners:
A. chemicallyinert liquid oils (Vaseline oil)
B. synthetic disaccharides(Lactulose, Sorbitol).
C. glycerin suppositories
LAXATIVES](https://image.slidesharecdn.com/pharmalectures-240325162433-8ad609f1/85/pharma-lectures-pdfMBBS-3rd-year-course-all-topics-276-320.jpg)