Downloaded 24 times



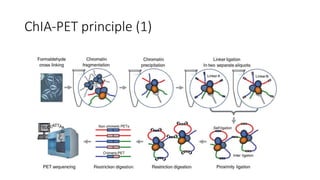
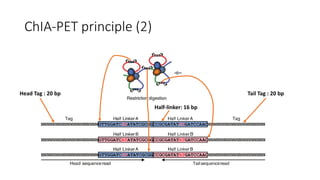
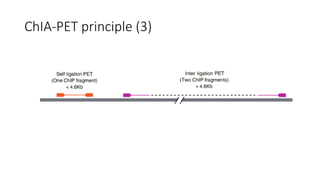

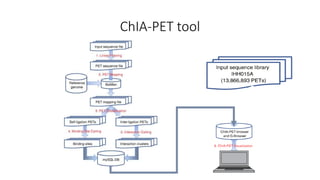

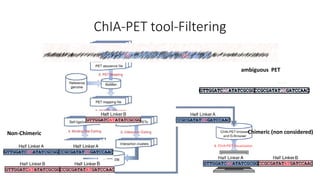
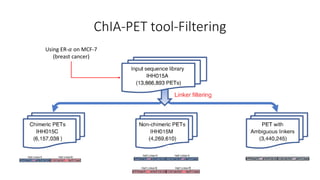
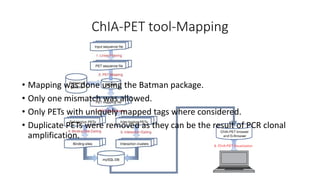
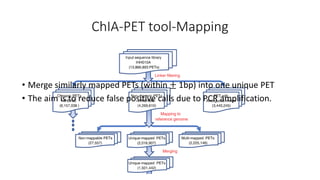
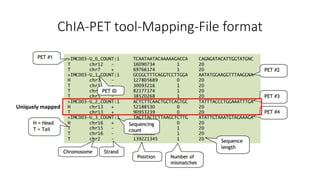
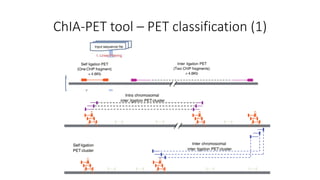


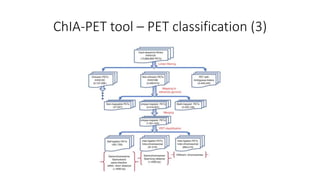
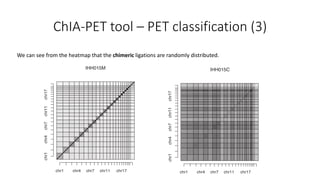
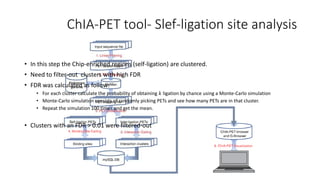
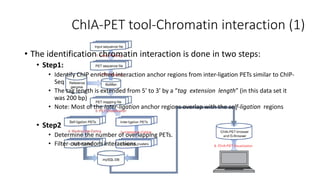
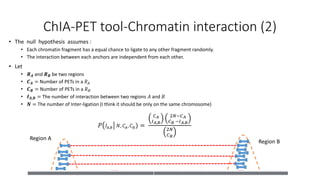


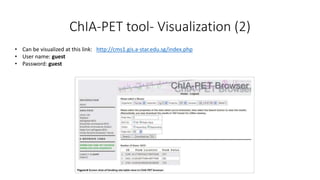




This document outlines the principles and implementation challenges of ChIA-PET analysis pipelines. It describes how ChIA-PET data is filtered, mapped, and classified into self-ligation and inter-ligation PETs. Significant chromatin interactions are identified using a statistical approach accounting for random interactions. Visualization of interactions is enabled through a genome browser. Instructions are provided for installing ChIA-PET analysis tools and running the pipeline on a local server.

















































![ANIMAL_CELL_,_TISSUE_AND_ORGAN_CULTURE[1].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/animalcelltissueandorganculture1-260204172026-4462b440-thumbnail.jpg?width=640&height=640&fit=bounds)





