INTRODUCTION
The vastmajority of microbial species remain
uncultivated and, until recently, about half of
all known bacterial phyla were identified only
from their 16S ribosomal RNA gene
sequence.
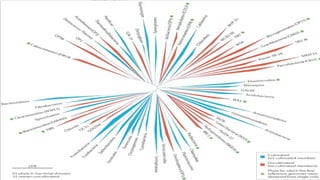
With the advent of single-cell sequencing,
genomes of uncultivated species are rapidly
filling in unsequenced branches of the
microbial phylogenetic tree.
3.
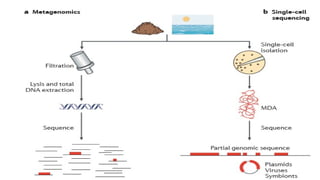
Sequencing DNAfrom single cells has opened
new windows onto the microbial world.
The use of single-cell sequencing in
combination with metagenomic analysis is
also emerging as a powerful new strategy to
analyse bacterial communities.
4.
THE UNCULTURED MAJORITY
The presence of highly conserved regions in the 16S
rRNA gene allows the design of PCR primers that can
amplify this gene from most bacteria with no need to
culture them (Giavanonni et al.,1991).
A second breakthrough was the development of
shotgun sequencing of DNA extracted from
environmental samples — a strategy called
metagenomics ( Handelsman et al.,2004).
5.
In onemetagenomic investigation, the Global Ocean Sampling
study (GOS study), the genes encoding 6 million new proteins were
sequenced, nearly equaling the total number of all previously
known proteins (Yooseph,S.,et al,2007).
However, in such studies, sequence data are obtained from the
millions of organisms present in each sample, and it has generally
not been possible to assemble the genomes of individual species,
except for some of the most abundant in the community (Gilbert,
J.A & Dupont ,2011).
6.
Sequencing fromsingle cells offers an alternative approach to
obtaining the genomes of individual organisms.
To obtain sufficient DNA template, it is necessary to amplify the
minute amounts of genomic DNA present in a single cell using
whole-genome amplification methods.
8.
THE DEVELOPMENT OFMDA
Whole-genome amplification from a single cell was first achieved in
pioneering work involving randomly primed PCR(Zhang,L.,et al,1992).
However, the thermal cycling involved made the resulting sequence
coverage highly biased, and limited the amplification products to
several hundred base pairs.
In 1991, a method was described for isothermal amplification in which
random primers are extended by the Klenow fragment at 37 °C
(Hartley J.L,1989).
The polymerase can copy the same region of a template more than
once by using a strand displacement mechanism, in which the
polymerase extends upstream primers while displacing the older
products of downstream priming.
9.
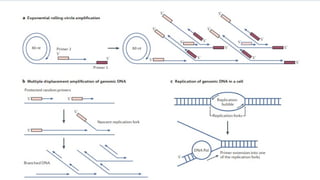
Another advancecame with the amplification of small circular
DNA templates20 using the φ29 DNA polymerase21, which has
greater processivity and stronger strand displacement activity
than the Klenow fragment ( Blanco, L et al.,1989).
This remarkable DNA polymerase forms a tight complex with
DNA, with a half-life of 1–13 minutes (depending on the
reaction conditions), and can invade double-stranded DNA
templates, thus initiating strand displacement (Hosono S, et
al.,2003).
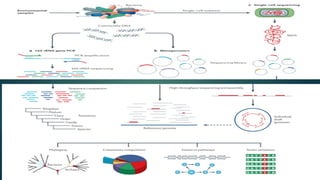
Multiple displacement amplification (MDA), allows enormous
amplification of any DNA template( Dean F.B, et al.,2002).
10.
MDA issimilar to cellular DNA replication in which the DNA
polymerase invades a replication fork and the large DNA products
that can be obtained (12 kb average, but ranging up to >100 kb)
are suitable for many biotechnological applications.
11.
WHOLE GENOME AMPLIFICATION
29 polymerase enzyme is already used in multiple displacement amplification (MDA)
procedures. The enzyme has many desirable properties that make it appropriate for
whole genome amplification (WGA) by this method.
High processivity.
Generates large fragments, over 10kb.
Produces more DNA than PCR-based methods, by about an order of magnitude.[8]
Requires minimal amount of template.
Novel replication mechanism; multiple-strand displacement amplification.
Random primers (hexamers) can be used, no need to design specific primers/target
specific regions.
No need for thermal cycling.
Good coverage and a reduced amplification bias when compared to PCR-based
approaches. There is speculation that it is the least biased of the WGA methods in use
15.
Applications
Sequencing ofmarine bacteria and single-cell eukaryotes has
provided new insights into both the structure of complex
microbial communities and the physiology of microorganisms.
Uncultured microorganisms have been targeted as new
sources of therapeutic agents and novel enzymes for industrial
use.
Recent single-cell studies of the human microbiome also
promise to advance our understanding of the microbial
communities that affect human health.
16.
References
Roger S.lasken,Jeffrey S.Mclean. Recent advances in genomic DNA
sequencing of microbial species from single cells. Nat Rev Genet.
15(9):577-584(2014).
Roger S. lasken , Genomic sequence of uncultured microorganisms
from single cells. Nat Rev Genet. Vol 10 (2012).
Raghunathan, A. et al. Genomic DNA amplification from a single
bacterium. Appl. Environ. Microbiol. 71, 3342–3347 (2005).
Lasken, R. S. in Whole Genome Amplification: Methods Express Series
(eds Hughes, S. & Lasken, R.) 99–118 (Scion, 2005).
Handelsman, J. Metagenomics: applications of genomics to
uncultured microorganisms. Microbiol. Mol. Biol. Rev. 68, 669–685
(2004).









![WHOLE GENOME AMPLIFICATION
29 polymerase enzyme is already used in multiple displacement amplification (MDA)
procedures. The enzyme has many desirable properties that make it appropriate for
whole genome amplification (WGA) by this method.
High processivity.
Generates large fragments, over 10kb.
Produces more DNA than PCR-based methods, by about an order of magnitude.[8]
Requires minimal amount of template.
Novel replication mechanism; multiple-strand displacement amplification.
Random primers (hexamers) can be used, no need to design specific primers/target
specific regions.
No need for thermal cycling.
Good coverage and a reduced amplification bias when compared to PCR-based
approaches. There is speculation that it is the least biased of the WGA methods in use](https://image.slidesharecdn.com/dnasequencinginclassification-250413054814-4191773c/85/Dna-sequencing-in-classification-_microb-11-320.jpg)