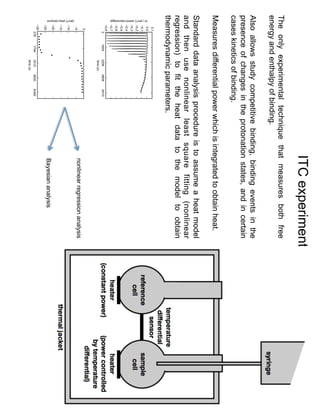
1. David Minh studies constrained molecular dynamics simulations, protein-ligand binding free energies using multiple rigid receptor structures, and Bayesian analysis of isothermal titration calorimetry data.
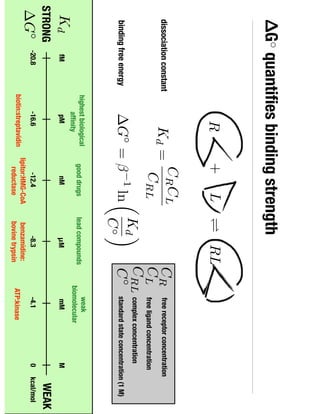
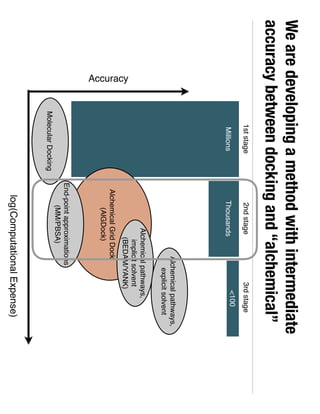
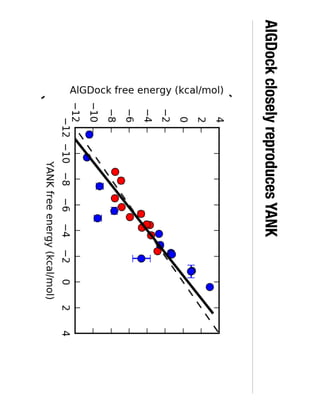
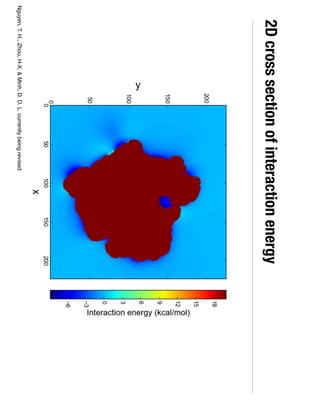
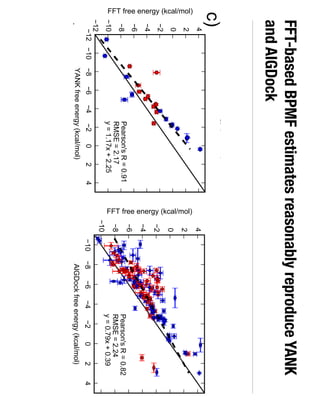
2. Binding free energy quantifies binding strength and can be estimated using methods like alchemical free energy calculations, molecular docking, or a new method called alchemical grid docking which uses binding potential of mean force estimates.
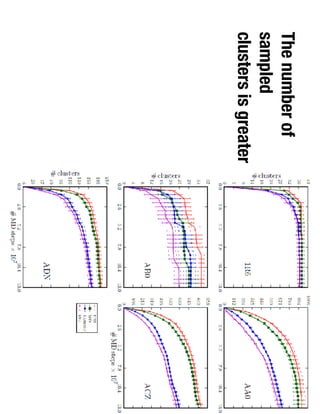
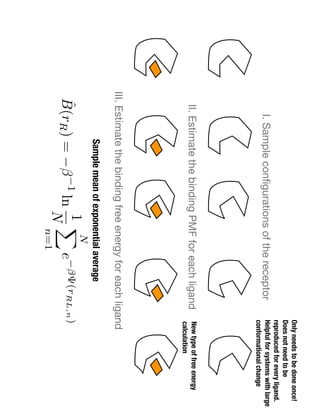
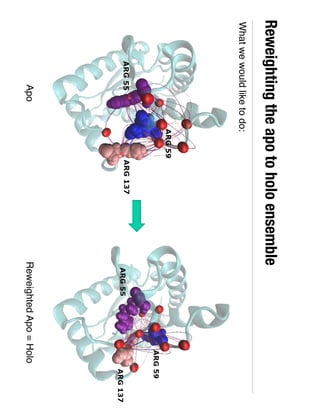
3. Reweighting configurations from apo protein ensemble simulations using binding potential of mean forces can reproduce holo protein ensembles, validating this new type of free energy calculation method.









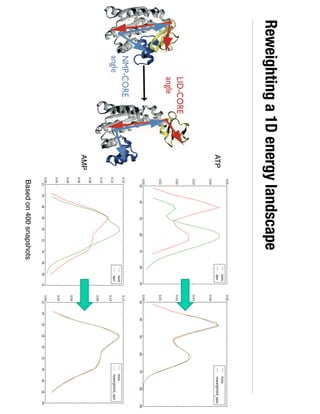

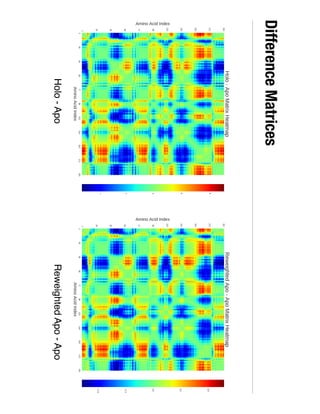
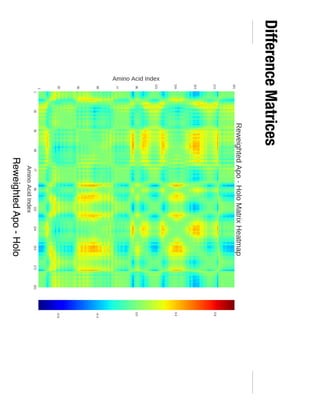


























































![Polymer [ बहुलक ] Chemistry Notes PDF - Irfanullah Mehar - JJ Sir Chemistry.pdf](https://cdn.slidesharecdn.com/ss_thumbnails/polymerchemistrynotespdf-irfanullahmehar-jjsirchemistry-260210172118-3f9b37f7-thumbnail.jpg?width=640&height=640&fit=bounds)


















