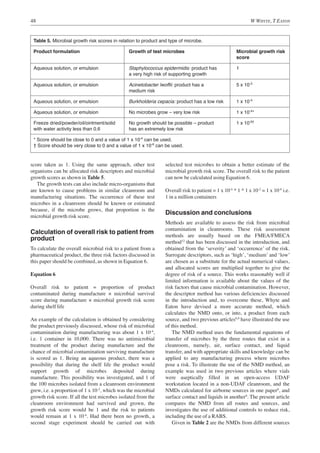
This document discusses methods for assessing the degree of risk from microbial contamination in cleanrooms. It describes using the number of microbes deposited (NMD) onto a product to calculate risk from airborne, surface contact, and liquid sources of contamination. The NMD method is demonstrated using a vial filling process in a cleanroom workstation. The document also considers the risk to patients from any microbes deposited in a product surviving during shelf life.