• Blood isa liquid tissue consisting of plasma
and cells.
• The blood cells include
– Red blood cells
– White blood cells
– Platelets
3.
• The redblood cells are also called erythrocytes.
• They are the most abundant blood cells.
• They contain haemoglobin (which carries
oxygen from lungs to tissues) and carbonic
anhydrase.
– Transport oxygen
– Acid-base buffer
– Bicarbonate transport
4.
• Basic characteristicsof red blood cells
– They are biconcave disks
– They can alter their shape as they squeeze
through capillaries.
– In men, the average number of RBCs is 5,200,000
(±300,000); in women, 4,700,000 (±300,000)/cubic
mm.
– Maximum amount of haemoglobin is 34g/100 mls
of cells.
5.
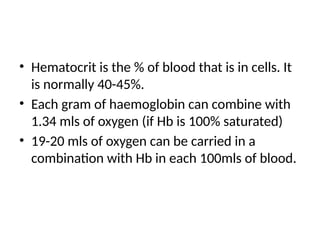
• Hematocrit isthe % of blood that is in cells. It
is normally 40-45%.
• Each gram of haemoglobin can combine with
1.34 mls of oxygen (if Hb is 100% saturated)
• 19-20 mls of oxygen can be carried in a
combination with Hb in each 100mls of blood.
6.
• Production ofred blood cells
– Yolk sac: during early weeks of embryonic life
– Liver: main site of red blood cell production during
mid trimester. Assisted by spleen and lymph
nodes.
– Bone marrow: exclusively produce red blood cells
during the last month of gestation and after birth.
7.
• In anadult >20 years old, the production of
red blood cells is mainly by the marrow of the
membranous bones, such as the vertebrae,
sternum, ribs and ilia.
• The marrow of all bones produces red blood
cells until a person is 5 years old.
8.
• Formation ofred blood cells
– All blood cells originate from a single type of cell
called the pluripotential haematopoeitic stem cells.
– PHSC → CFU-S→CFU-B→CFU-B→ CFU-E→
Erythrocytes
– CFU-E →Proerythroblast→ basophil erythroblast→
polychromatophil erythroblast→ orthochromatic
erythroblast→ reticulocyte→ erythrocyte
9.
• The concentrationof reticulocyte is < 1%.
• Growth and reproduction of the different stem
cells are controlled by multiple proteins called
growth inducers like IL-3.
• Formation of these inducers may be
controlled by factors such as hypoxia for RBCs
and infections for WBCs.
10.
• Role oferythropoeitin
– Erythropoeitin (EPO) stimulates red blood cells
– EPO is a glycoprotein hormone with a molecular weight of
34,000
– 90% of erythropoeitin is formed in the kidneys and 10% in
the liver.
– EPO is produced in the kidneys by the peritubular
fibroblasts (mainly) and by the renal epithelial cells
– EPO stimulate the production of proeryhtroblasts from
haematopoeitic stem cells in the bone marrow and speeds
up production of new red blood cells.
11.
– In theabsence of EPO, few red blood cells are
formed by the bone marrow.
– Tissue hypoxia is the most important stimuli for
EPO production
– Factors causing tissue hypoxia include high
altitude, anaemia, bone marrow loss, chronic lung
disease, chronic heart failure.
– Other stimuli for EPO production: norepinephrine,
epinephrine, prostaglandins.
12.
• Mechanism:
– Renaltissue hypoxia leads to increased expression
of a transcription factor called hypoxia- inducible
factor-1 (HIF-1) which codes for the EPO gene and
other hypoxia-inducible gene.
– HIF-1 induces transcription of mRNA in the EPO
gene and so increase EPO production.
13.
• Role ofvitamin B12 and folic acid
– Both vitamins are essential for the final
maturation of red blood cells - synthesis of
thymidine triphosphate (a building block of DNA).
– Deficiency of one or both vitamins lead to failure
of nuclear maturation and cell division leading to
production of megaloblasts (macrocyte with
irregular, large nuclei)- Megaloblatic anaemia( a
type of macrocytic anaemia)
14.
• Vitamin B12is absorbed from the terminal
ileum
• It is stored in large quantities in the liver
(about 1,000 micrograms)
• It is slowly released as needed by the bone
marrow
• 1-3 micrograms are needed per day.
• Liver stores take 3-4 years to be depleted.
15.
• Pernicious Anaemia
–Results form failure to absorb vitamin B12 from
the gut due to intrinsic factor deficiency secondary
to atrophic gastritis.
– Intrinsic factor combines with vitamin B12 in food
and protects it from digestion by gut secretions
– Intrinsic factor binds to receptors in the terminal
ileum and vitamin B12 is transported into the
blood by pinocytosis.
16.
• Pteroylglutamic aciddeficiency
– This describes failure of red blood cell maturation
secondary to folic acid deficiency alone.
– Folic acid is found in green vegetables, fruits and
meats.
– Causes
• Sprue
• Lack of dietary folate
17.
• Haemoglobin formation
–Synthesis begins in the proerythroblasts and
continues up to the reticulocyte stage.
– Succinyl Co A + glycine → pyrole molecule (4) →
protoporphyrin IX + iron → Heme
– Heme + globin (polypetide) →haemoglobin chain,
mw16,000 (α or β chains).
– 2α
and 2β chains form Haemoglobin A (HbA)
18.
• Each haemoglobinchain binds one molecule
of oxygen, therefore an Hb molecule with 4
chains will bind 4 atoms of oxygen.
• Abnormality of the chains can alter the
physical characteristics of the Hb molecule.
• Hb reversibly binds with oxygen (transported
as molecular oxygen rather than oxygen ion)
and is readily released into the tissues.
19.
• Sickle cellanaemia-
– Characterized by an abnormality in haemoglobin
chain in which valine replaces glutamic acid at
position 6 of the β-globin chain.
– This HbS tends to crystallize at low oxygen tension
and are easily destroyed leading to sickle cell
anaemia.
20.
• Iron metabolism
–Iron is important for the formation of Hb,
myoglobin, cytochromes, cytochrome oxidase,
peroxidase, catalase.
– Total body iron is about 4-5g
– 65% is in the form of haemoglobin, 4% in the form
of myoglobin, 1% in the form of various heme
compounds (taking part in intracellular oxidation),
0.1% is combined with transferrrin, 15-30% is
ferrritin
21.
• Ferritin ismainly in the liver parenchymal
cells. Some ferritin is also found in other cells
of the reticuloendothelial system.
• Iron absorbed from the small intestine,
combines with apotransferrin to form
transferrin.
• Transferrin is transported in the plasma and is
easily released to any body tissue.
22.
• In thecell cytoplasm, iron combines with apoferritin
to form ferritin (storage iron).
• Smaller quantities of the iron in the storage pool are
in an extremely insoluble form called haemosiderin
( when iron is in excess).
• When iron concentration is low, ferritin releases iron
in the circulation.
• Failure to transport iron to the erythroblasts results in
the formation of sideroblasts – sideroblastic anaemia.
23.
• Iron absorptionis slow, therefore only small
amounts are absorbed irrespective of dose.
• When all iron stores are filled, the rate of
additional iron absorption from the intestinal
tract becomes greatly decreased.
• When iron stores are depleted, the rate of
absorption can accelerate 5 times or more.
• Total body iron is regulated mainly by altering
the rate of absorption.
24.
• Iron isexcreted mainly via faeces, about 0.6
mg per day for males. For females, 1.3 mg/day
including menstrual loss.
25.
• Red bloodcells have cytoplasmic enzymes
whose functions include:
– Metabolize glucose to produce ATP
– Maintain pliability of cell membrane
– Maintain membrane transport of cell membrane
– Keep iron in Hb in ferrous form rather than ferric
form
– Prevent oxidation of proteins in red blood cells
26.
• The averagelifespan of red blood cells is about
120 days.
• When the red blood cells (8 um) gets old, they
become fragile and are destroyed as they go
throught the red pulp of the spleen (3um).
• Once destroyed, Hb is released and is
phagocytized by body macrophages.
• The macrophages release the iron from Hb into
the blood.
27.
• This iscarried by transferrin either to the bone
marow or to the liver.
• The porphyrin portion is converted by the
macrophages into the bile pigment bilirubin.
28.
• Bilirubin Metabolism:
–Hb is split into heme and globin
– Heme ring is opened to give free iron and a straight
chain of pyrrole nucei from which bilrubin is formed.
– Bilirubin is a major pigment in bile
– Biliverdin→ free bilirubin (unconjugated bilirubin)→
conjugated bilirubin
– Unconjugated bilirubin is strongly bound to plasma
albumin in the blood.
29.
• In theliver, it is conjugated with glucoronic
acid and sulphate.
• Conjugated bilirubin is excreted from the
hepatocytes by an active transport process
into the bile canaliculi and then into the
intestines.
• In the intestine, 50% conjugated bilirubin is
converted to urobilinogen via bacterial action.
30.
• Urobilinogen isreabsorbed through the
intestinal mucosa into the blood and most is
re-excreted by the liver back into the gut
(entero-hepatic circulation)
• 5% is excreted by the kidneys into the urine
• After exposure to air, the urobilinogen in urine
is converted to urobilin (in urine) and
stercobilin (in faeces).
31.
• Jaundice
– Definedas the yellowish discolouration of the skin
and mucous membrane.
– Due to excess bilirubin in the extracellular fluid.
– Common causes include:
• Haemolysis
• Obstruction of bile ducts or damage to the liver cells. It
is characterised by pale stools, bilirubinuria and
absence of urobilinogen in urine.
32.
ANAEMIAS
• Anaemia isdefined as a decreased in Hb or a
reduction in red blood cell mass.
• Types of anaemia
– Microcytic hypochromic anaemia
– Aplastic anaemia
– Megaloblastic anaemia
– Haemolytic anaemia
– Blood loss anaemia
33.
• Microcytic hypochromicanaemia may be due
to:
– Iron deficiency
– Lead poisoning
– Sideroblastic anaemia
– Thalassemia minor
34.
• Aplastic anaemiamay be due to
– Radiation therapy/chemotherapy
– Exposure to high dose toxins like hydrocarbons or
insecticides.
– Autoimmune disorders like SLE
– Viral infections like HIV, Parvovirus B-19.
– Drugs: chloramphenicol, methothrexate,
busulphan, cyclophosphamide
– Idiopathic
35.
• Megaloblastic anaemia:Due to folate or
vitamin-B12 deficiency. Causes include:
– Pernicious anaemia
– Total gastrectomy
– Sprue
36.
• Haemolytic membrane:Due to abnormal red
blood cell membrane or haemoglobin.
• Common causes include
– Hereditary spherocytosis
– Sickle cell anaemia
– Erythroblastosis fetalis: A condition in which Rh+
red blood cells in the fetus are attacked by
antibodies from a Rh- mother. This leads to
congenital anemia.
37.
• Effects ofanaemia on function of circulatory
system:
– Blood viscosity falls
– Total peripheral resistance falls: this leads to
increased venous return and cardiac output.
– Increased cardiac work load
– In the face of exercise, extreme tissue hypoxia
results and acute cardiac failure may ensue.
38.
POLYCYTHAEMIA
• Defined asred blood cell count of at least 30%
above normal or 6-7 million/mm3; PCV 60-
70%.
• Secondary polycythaemia – secondary to
tissue hypoxia
• Causes of secondary polycythaemia include
– High altitude (physiological polycythaemia)
– Failure of oxygen delivery to the tissues
39.
• Polycythaemia vera(erythemia)
– A pathological condition characterized by red
blood cell hyperproliferation. It is due to genetic
aberration in the haemocytoblastic cells that
produce the blood cells. This leads to excess
production of red blood cells; white blood cells
and platelets are also increased.
– Total blood volume increases, blood is
hyperviscous.
40.
• Effects ofpolycythaemia on function of the
circulatory system
– Sluggish blood flow through peripheral blood vessels
– Blood viscosity increases leading to decrease venous
return to the heart
– Blood pressure is normal or elevated
– Cardiac output is normal
– Ruddy complexion with a bluish(cyanotic) tint to the
skin- facial plethora
41.
PLATELETS
• They arederived from megakaryocytes formed
in the bone marrow.
• Mean number of platelets: 300,000/ml of blood
• Are small fragments, important in the initiation
of blood clotting.
• They are replaced once in every 10 days
• 30,000 platelets formed each day per ml of
blood.
42.
• They are1-4 um in diameter
• They lack nuclei and cannot reproduce
• Half life is 8-10 days
• They contain contractile proteins like actin,
myosin and thrombosthenin in their cytoplasm.
• They contain residual endoplasmic reticulum
and golgi apparatus that synthesize enzymes
and also store large quantities of calcium ions.
43.
• They produceATP, ADP, prostaglandins and
fibrin-stabilizing factor.
• They also contain large amounts of
phospholipids that can activate the blood
clotting process.
44.
WHITE BLOOD CELLS
•The white blood cells forms the body defense
to infectious agents like bacteria, fungi, viruses
and parasites.
• They work by phagocytosis or by forming
antibodies.
• Leukocytes include
– Granulocytes and monocytes (formed in blood)
– Lymphocytes and plasma cells (formed in lymph
tissue)
45.
• Granulocytes
– Arealso called polymorphonuclear cells. They
have multiple nuclei.
– Have a granular appearance
– Neutrophils, basophils and eosinophils.
46.
• The meanconcentration of white blood cells is
about 7,000 cells/min.
– 62% neutrophils
– 2.3% eosinophils
– 0.4% basophils
– 5.3% monocytes
– 30% lymphocytes
47.
• There are2 major white blood cell lineages:
myelocytic and lymphocytic lineages.
• Myeloblast→ promyelocyte → megakaryocytes
→ neutrophil myelocyte→ neutrophil
metamyelocyte → band neutrophils →mature
neutrophils
• Eosinophil myelocyte→ eosinophil
metamyelocyte→ mature eosinophil
• Basophil myelocyte → mature basophil
48.
• Granulocytes andmonocytes are formed only
in the bone marrow and are stored in the
marrow until needed by the circulatory system.
• Lymphocytes and plasma cells are formed in
lymphogenous tissues like lymph glands,
spleen, thymus, tonsils, pockets of lymphoid
tissue in the bone marrow or gut wall (peyer’s
patches).
49.
• Lymphocytes aremostly stored in various
lymphoid tissues, a small number is in the
blood.
• The lifespan of granulocytes after being
released from the bone marrow is 4-8 hours in
blood and 4-5 days in tissues.
• Monocytes have a short transit time of 10-20
hours in blood before entering the tissues.
50.
• Lymphocytes enterthe circulatory system
continually, along with drainage of lymph from
lymph nodes and other lymphoid tissue.
• After a few hours, they pass out of the blood
back into the tissues by diapedesis, re-enter
the lymph and return to the blood again.
51.
Neutrophils and Macrophages
•Neutrophils can attack and destroy an invading
organism in the blood.
• Macrophages begin life as blood monocytes.
• Neutrophils and monocytes can squeez through
the pores of the blood capillaries by diapedesis
and move through tissue planes by ameboid
motion.
• They move toward an invading organism by
chemotaxis.
52.
• Stimuli forchemotaxis include:
– Bacterial or viral toxin
– Degenerative product of inflamed tissue
– Products of the complement system
– Fibrin-degradation products
53.
Phagocytosis
• Described asthe cellular ingestion of the
offending agent
• Most important function of neutrophils and
macrophages
• The process by which a pathogen is selected
for phagocytosis and destruction is called
opsonization.
54.
• Selective factorsthat favour phagocytosis
– Rough surface of infected tissues
– Loss of protective coat as seen in dead tissues and
foreign particles
– Presence of antibodies that adhere to the
bacterial membranes making it susceptible to
phagocytosis. Antibodies attach to C3 which inturn
attach to receptors on the phagocyte membrane,
thus initiating phagocytosis.
55.
• Phagocytosis byneutrophils
– Neutrophil attaches itself to the particle and then
projects pseudopodia in all directions around the
particle.
– The pseudopodia meet one another on the
opposite side and fuse, forming an enclosed
chamber containing the phagocytized particle.
– The chamber invaginates and breaks away from
the outer cell membrane to form a phagosome.
56.
• Phagocytosis byneutrophils
– A phagosome is a free-floating phagocytic vesicle
– A single neutrophil can usually phagocytize 3-20
bacteria before the neutrophil becomes
inactivated and dies.
57.
• Phagocytosis bymacrophages
– They are more powerful phagocytes than
neutrophils
– May phagocytise up to 100 bacteria
– They can engulf much larger particles like malaria
parasites, red blood cells.
– After digesting the particles, macrophages can
extrude the residual products and may survive and
function for many months.
58.
• Lysosomes andother cytoplasmic granules in the
neutrophil or macrophage
– When in contact with the phagocytic vesicle, they fuse with
the vesicle and release their digestive enzymes and
bactericidal agents into the vesicle transforming it to a
digestive vesicle.
– In macrophages, the lysosomes also contain lipases which can
digest thick lipid membranes found in organisms like M.TB.
– Bactericidal agents include superoxide, hydrogen peroxide,
hydroxyl ions
– Lysosomal enzymes include myeloperoxidase,
59.
Reticuloendothelial system
• Alsocalled monocyte-macrophage cell system.
• Monocytes enter the tissues and become
macrophages.
• The tissue macrophages may be mobile or fixed.
• Both groups maintain their phagocytic activity
• The fixed macrophages may attach to tissues for
months/years until they are called upon to
perform specific local protective function.
60.
• The totalcombination of monocytes, mobile
and fixed macrophages, specialized endothelial
cells in the bone marrow, spleen and lymph
nodes is called the reticuloendothelial system.
• Examples of such macrophages: histiocytes,
kupffer cells, microglia, alveolar macrophages,
macrophages in lymph nodes, spleen and bone
marrow.
61.
Inflammation
• Is definedas tissue response to injury.
• Evidenced by rubor(redness),calor (heat),
dulor(pain), tumor(swelling), loss of function.
• Mediators-
– histamine, bradykinin, serotonin, prostaglandins,
lymphokines
– Products of the complement system and blood
clotting system.
62.
• Inflammation ischaracterized by
– Vasodilation of local vessels
– Increased capillary permeability
– Increased formation of fibrinogen and other
proteins leaking from capillaries leading to clotting
of the fluid in the interstitial spaces.
– Migration of granulocytes and monocytes into the
tissue.
– Tissue swelling
63.
• Walling-off effect
–The tissue spaces and the lymphatics in the
inflamed area are blocked by fibrin clots so that
fluid barely flow through the spaces
– This walling-off process delays the spread of
bacteria or toxic products.
64.
• Role ofmacrophages and neutrophils in
inflammation:
– Tissue macrophages provide a first line of defense
against infection
– Neutrophil invasion of the inflamed area is a second
line of defense
– Second macrophage invasion into the inflamed tissue
is a third line of defense
– Increased production of granulocytes and monocytes
by the bone marrowis a fourth line of defense.
65.
• Neutrophils areattracted to an inflamed tissue
by cytokines such as TNF and IL-1 through the
following processes:
– Margination: The neutrophils stick to the capillary
and venule walls in the inflamed area.
– Diapedesis: The intercellular attachments between
endothelial cells is made loose and neutrophils can
move into tissue spaces.
– Chemotaxis: neutrophils move toward injured tissue.
66.
• Neutrophilia: Increasein the number of
circulating neutrophils in the face of infection
or inflammation.
– The products of inflammation acts on the bone
marrow to mobilize stored neutrophils.
• Neutropenia: reduced number of neutrophils
in the blood.
67.
• The mainfactors involved in the feedback
control of macrophage and neutrophil
responses are TNF, IL-1, GM-CSF, G-CSF, M-CSF.
• These factors are produced by macrophages in
the inflamed tissue.
• Pus contains necrotic tissue, dead neutrophils
and macrophages, tissue fluid. It autolyzzes
over a period of days.
68.
• Eosinophils
– 2%of blood leukocytes
– Weak phagocytes
– Exhibit chemotaxis
– Produced inlarge numbers by people with parasitic
infections and allergic responses.
– They are granular
69.
• Eosinophils
– Theyattach to parasites by special surface
molecules and kill the parasites.
– They release hydrolytic enzymes (modified
lysosomes), reactive oxygen species and major
basophilic protein (a larvicidal polypeptide)
– The mast cells and basophils in allergic reactions
release release an eosinophil chemotactic factor
that causes eosinophils to migrate toward the
inflamed allergic tissue.
70.
• Basophils
– Similarto large tissue mast cells
– Liberate heparin into the blood
– They also release histamine, bradykinin, serotonin
– They play an important role in allergic reactions
71.
• Leukopenia
– Decreasedproduction of white blood cells by the
bone marrow.
– Allows normal flora to invade adjacent tissues
– Clinical features include: mouth ulcers, diarrhoea,
reccurent severe respiratory infections.
– Causes include: irradiation, drugs, chemicals
72.
• Leukaemias
– Characterisedby increased numbers of abnormal
white blood cells (blast cells) in the circulating
blood.
– Due to cancerous mutation of a myelogenous or
lymphogenous cells.
– These abnormal cells are produced in the bone
marrow and extramedullary tisues such as lymph
nodes, spleen and liver.
73.
• Common leukemicfeatures include
– Development of infection
– Severe anaemia
– Bleeding tendency
– Protein-wasting
74.
IMMUNOLOGY
• The abilityof the body to resist invading
organism is called immunity
• Immunity may be innate or acquired.
• Innate immunity is non-specific and it involves
the following:
– Phagocytosis, acid in stomach, resistance of skin
to invading organisms, lysozyme, basic
polypeptide, complement system, natural killer
cells.
75.
ACQUIRED IMMUNITY
• AcquiredImmunity: A powerful, specific
immunity against individual invading agents.
• It involves formation of antibodies or activated
lymphocytes.
• May be divided into:
– Humoral or B cell immunity
– Cell-mediated/ T-cell immunity
76.
• Antigens: Theseare large proteins or
polysaccharides that initiate the acquired
immune response.
• Antigens possess
– High molecular weight
– Presence of epitopes on their surface
77.
• Lymphocytes
– Asubset of white blood cells that are located
mostly in the lymph nodes, spleen, thymus,
submucosal areas of the GI tract, bonne marrow,
adenoids, tonsils.
– They are distributed in the body to intercept
invading organisms or toxins
– Two major populations of lymphocytes: T-
lymphocytes and B-lymphocytes.
78.
• Both arederived from haematopoeitic stem cells.
• The T-lymphocytes are preprocessed in the
thymus gland.
• The B lymphocytes were first discovered in a
preprocessing organ in birds called bursa of
fabricus (B- lymphocytes)
• B-lymphocytes are preprocessed in the liver
during mid fetal life and in bone marrow in late
fetal life and after birth.
79.
• In thethymus, the T lymphocytes acquire
specific reactivity against one antigen and the
abilty to recognise self [self tolerance].
• The preprocessing of T lymphocytes in the
thymus occurs shortly before birth and a few
months after birth.
• After preprocessing, the B lymphocytes migrate
to lymphoid tisue throughout the body where
they lodge near the T lymphocyte areas.
80.
• When specificantigens come in contact with T
and B- lymphocytes in the lymphoid tissue, the
T lymphocytes become activated to form
activated T cells and the B lymphocytes
become activated to produce antibodies.
• These activated T cells and antibodies, in turn,
react highly specifically against particular types
of antigens that initiated their development.
81.
• All thedifferent lymphocytes that are capable of
forming one specific antibody or T cell are called a
clone of lymphocytes.
• The clone cells are:
– Similar
– Derived from a one or few early progenitor cells.
• In the stem cells, there are gene segments instead
of whole genes for forming clones of lymphocytes.
82.
• Each cloneof lymphocytes is responsive to
only a single type of antigen or to similar
antigens that have almost exactly the same
stereochemical characteristics.
– B cells have on its surface membrane about
100,000 antibody molecules that will react highly
specifically with only one type of antigen.
– T cells have surface markers or receptor proteins
which are highly specific for a particular antigen.
83.
• An antigentherefore stimulates only those
cells that have complementary receptors for
the antigen and are already committed to
respond to it.
84.
• Role ofmacrophages
– They act as antigen presenting cells
• This is done by cell-cell contact directly to the
lymphocytes thus leading to activation of the specified
lymphocytic clones.
– They secrete interleukin-1
• IL-1 promotes growth and reproduction of specific B
lymphocytes.
85.
• Role ofT cells
– Most antigens activate both T and B lymphocytes
at the same time.
– The T helper cells secrete secrete substances
called lymphokines that activate specific B
lymphocytes.
86.
Humoral Immunity
• Macrophagesin the lymphoid tissue present the
antigen to adjacent B and T lymphocytes.
• A subset of T cells (T helper cells) also serve as antigen
presenting cells to cause extreme B cell activation.
• The specific B lymphocytes for the antigen enlarge
and become lymphoblasts→ plasmablasts→ plasma
cells
• The mature plasma cells produce the antibodies
(gamma globulins).
87.
• A fewof the lymphoblasts do not form plasma cells
but go on to form new B lymphocytes similar to the
original clone. These lymphocytes are called
memory cells.
• Subsequent exposure to the same antigen will
cause a more rapid and potent antibody response.
• The increased potency and duration of the
secondary response explain why immunization is
usually given in multiple doses.
88.
• Nature ofantibodies:
– 20% of plasma proteins, Molecular weight
160,000-970,000.
– They are gamma globulins
– Consist of light and heavy polypeptide chains at
least (≥2 pairs) held together by covalent and non
covalent bonds.
– Each chain consist of a variable portion and a
constant portion.
89.
• Nature ofantibodies
– They exhibit specificity.
• Each antibody is specific for a particular antigen. An
antigen fits as a mirror image to the antibody.
90.
• Classes ofantibodies: IgM, IgG, IgA, IgD, Ig E.
– IgM is responsible for the acute response.
– IgG is responsible for the chronic response.
– IgE is responsible for allergies.
– IgA is attached to mucosal surfaces.
– IgD is coexpressed with IgM
91.
• Mechanism ofaction of antibodies
– Direct action on the invader. This effect may be
weak. It includes:
• Agglutination
• Precipitation
• Neutralization
• Lysis
– Activation of the complement system
92.
• Complement system:
–Consist of enzyme precursors
– Two major pathways: classical and alternate
pathway
• The classical pathway is initiated by antigen-antibody
reaction. An exposed portion of the antibody binds to
C1 molecule of the complement system setting into
motion a cascade of reactions.
• The alternate pathway is triggered when C3b protein
binds a microbe or foreign material or tissue.
93.
• Effects ofthe complement pathway
– Opsonization and phagocytosis: mediated by C3b
– Lysis: mediated by C5-9
– Agglutination
– Neutralization
– Chemotaxis: initiated by C5a
– Activation of mast cells and basophils: achieved by C3a, C4a
and C5a
– Increased local blood flow, increased capillary leakage of
proteins, coagulation of interstitial fluid proteins-
Inflammatory effects.
94.
Cell mediated immunity
•The T lymphocytes respond to antigens only
when they are bound to specific molecules
called MHC molecules on the surface of
antigen-presenting cells.
• The major antigen presenting cells are:
– Macrophages
– B lymphocytes
– Dendritic cells (most potent)
95.
• Binding ofT cell to antigen presenting cell is
mediated by cell adhesion molecules.
• The MHC proteins are encoded by a large group
of genes called the major histocompatibility
complex. The MHC bind peptide fragments of
antigen proteins. There are 2 types:
– MHC 1: present antigen to cytotoxic cells
– MHC 11: present antigen to T helper cells.
96.
• The antigenson the surface of antigen
presenting cells bind with receptor molecules
on the surfaces of T cells.
• Types of T cells
– T helper cells
– Cytotoxic T cells
– Suppressor T cells
97.
• T helpercells: most numerous T cells, major
regulators of immune function. They produce
lymphokines that act on other cells of the immune
system as well as on bone marrow cells 0– IL 2, IL 3,
IL4, IL5, IL6, GM-CSF, IFN-Y
• Cytotoxic T cells: They bind tightly to cells and
organisms and secrete perforins, allowing fluid to
flow rapidly into the cell from the interstitium. They
also release cytotoxic substances directly into the
attacked cell that dissolves the cell.
98.
• The cytotoxiccells play an important role in
destroying viruses, cancer cells and foreign
cells from transplantation.
• Suppressor T cells
– Are also called regulatory T cells
– They play a role in immune tolerance
– They are capable of suppressing the functions of
both cytotoxic and T helper cells.
99.
Immune Tolerance
• Theacquired immune response may destroy
one’s own tissues.
• This is prevented by self tolerance or immune
tolerance. The body recognizes it’s own tissues
as distinct from that of bacteria or viruses.
• However, few antibodies or activated T- cells
are formed against one’s antigens.
100.
• Self toleranceis due to preprocessing of T-
lymphocytes in the thymus and B-lymphocytes
in the bone marrow during which all or most
clones of lymphocytes specific to the body’s
tissues are destroyed.
• Failure of the tolerance mechanism leads to
autoimmune diseases- SLE, rheumatoid
arthritis, myasthenia gravis, GoodPastures
disease, post-streptococcal syndromes
101.
Vaccination
• Used toproduce acquired immunity against
specific tissues.
• Types
– Killed vaccines: typhoid fever, whooping cough,
diphtheria,
– Live attenuated vaccines: small pox, yellow fever,
poliomyelitis, measles,
– Recombinant vaccines: tetanus, botulism, HBV
102.
Passive immunity
• Involvesthe transfusion of antibodies or T-
lymphocytes to confer immunity.
• Transfused antibodies lasts for 2-3 weeks
• T-lymphocytes last for few hours to few days.
103.
Allergy/Hypersensitivity
• An untowardeffect of the immune system.
• First exposure is unremarkable, repeated
exposure cause the formation of activated helper
and cytotoxic T-cells leading to tissue damage.
• IgE antibodies are involved and they have a strong
tendency to attach to mast cells and basophils.
• An antigen reacts with a specific IgE antibody to
trigger an allergic reaction.
104.
• An IgE–antigen complex attach to membrane
of mast cells and basophils. These cells rupture
and release histamine, protease, SRSA,
eosinophil/neutrophil chemotactic substance,
heparin, PAF.
• This results in local blood vessel dilation,
increased capillary permeability, contraction of
smooth muscle cells, attraction of eosinophils
and neutrophils.
105.
• The clinicaleffects include
– Anaphylaxis- widespread activation
– Urticaria- limited to the skin
– Asthma – limited to the lungs
– Hay fever- upper airways
• Treatment: Antihistamines, steroids
106.
BLOOD TRANSFUSION
• Theblood of different people have different
antigenic and immune properties.
• This provides the basis for blood transfusion
reactions.
• Major types of antigens involved in blood
transfusion reaction: ABO system, Rhesus(Rh)
system.
107.
• ABO system(O-A-B System)
– Type A and type B antigen occur on the surfaces of the red
cells- Agglutinogens.
– The antibodies are called agglutinins
• Blood types
– Type A contain only type A agglutinogen, anti B agglutinins
– Type B contain type B agglutinogen, anti A agglutinins
– Type AB contain A and B agglutinogen; no aagglutinins
– Type O lack A or B agglutinogen; anti A and anti B
agglutinins
108.
• AB –is the universal recipient
• O- is the universal donor
• Agglutinins are gamma globulins (IgG and IgM
produced by the bone marrow and lymph glands.
• At birth, the levels of agglutinin is zero,
production starts 2-8 months after birth and a
maximum titre is reached at 8-10 years of age
and begins to decline.
109.
• Small amountsof type A and B antigens enter
the body through food, in bacteria and in
other unspecified ways. These substances
initiate the development of the anti A and anti
B agglutinins.
110.
• Transfusion reactions
–When blood cells are mismatched, they agglutinate as a
result of the agglutinins attaching themselves to the red
blood cells.
– Agglutinated cells plug small blood vessels and may
rupture leading to haemolysis.
– Transfusion reactions are spontaneous in ABO system but
delayed in the Rhesus system.
• Before blood transfusion, the blood type of the donor
and recipient should be appropriately matched.
111.
• Rhesus Bloodtype
– The rhesus antigens are also called rhesus factor.
– There are 6 common rhesus antigens: C,D,E,c,d,e
– Rhesus antigen type D is widely prevalent in the
population, is more antigenic than the others and is used
to define rhesus positivity.
– In the Rhesus system, spontaneous agglutinins almost
never occur. Instead, the person must first be exposed to a
rhesus antigen before enough agglutinins are made to
cause a significant transfusion reaction.
– Anybody with type D antigen is rhesus positive.
112.
• Rhesus BloodTransfusion
– First exposure: no immediate reaction
• However, anti-Rh antibodies can develop in sufficient
quantities during the next 2-4 weeks (reaching a
maximum in 2-4 months) to cause agglutination of
transfused red cells which are then haemolysed –
Delayed transfusion reaction.
– On second exposure, transfusion reaction is
immediate and severe, similar to ABO reactions.
113.
Erythroblastosis foetalis
• Erythroblastosisfoetalis
– Characterized by agglutination and phagocytosis of
foetal red cells.
– Mother is Rh-,Father Rh+, Baby inherits Rh+ from father
– At birth, the maternal and foetal blood are mixed,
exposing the mother to Rh+ antigens leading to
development of agglutinins.
– First baby is spared, incidence rises with subsequent
pregnancies as the anti Rh+ antibodies diffuse into
foetal blood at birth.
114.
– This leadsto haemolysis of foetal red blood cells and
neonatal jaundice and anemia.
– The antibodies can also attack and damage other
cells of the body.
– The antibodies may persist for more than a month
after birth, destroying more red blood cells leading
to hepatosplenomegaly and appearance of
circulatory blast cells.
– Severe permanent brain damage due to precipitation
of bilirubin in the neuronal cells – Kernicterus.
• Transfusion reactions
–Immediate
– Delayed
– Transfusion related acute lung injury
– Transfusion related acute kidney injury
117.
Tissue/Organ Transplantation
• Thered blood cell antigens that cause transfusion
reactions are present in other cells of the body.
• Thus, the mechanisms of blood transfusion
reactions matches that of tissue or organ
rejection.
• The most important antigens for causing graft
rejection are a complex called the human
leucocyte antigens (HLA) on leucocytes and other
tissues.
118.
• The transplantedtissue is called a graft.
• Types of graft:
– Autograft: from the same animal
– Isograft: from an identical twin to another.
– Allograft: from one human being to the other or
from one animal to the other of the same species.
– Xenograft: transplant from an animal to a human
being or from an animal of one species to one of
another species.
119.
• For autograftsand isografts, the cells in
transplant contain virtually the same types of
antigens as the recipient.
• For allografts and xenografts, immune
reactions occur causing death of cells except
specific therapy is instituted.
• Examples of allografts: kidney, lungs, liver,
heart, bone marrow.
120.
• To preventgraft rejection, the immune system
is suppressed with the following drugs:
– Glucocorticoids
– Azathioprine
– Calcineurin Inhibitors
– ATG/ALG
• Major adverse effect of immunosuppressives
are increased incidence of infections and
malignancies.
121.
HAEMOSTASIS
• Haemostasis isthe prevention of blood loss.
• The mechanisms involved in haemostasis:
– Vasoconstriction
– Platelet-plug formation
– Blood clot formation
– Growth of fibrous tissue into the blood clot.
122.
• Vasoconstriction: Followingdamage to a blood
vessel, the smooth muscle in the wall contract
immediately to reduce the flow of blood from
the damaged vessel.
• The mechanisms involved:
– Local myogenic spasm and neural reflexes initiated
by pain.
– Local factors from damaged tissues and blood
platelets like thromboxane A2.
123.
• Platelet plug:
–The platelet cell membrane contain glycoproteins
and phospholipids.
– The platelet glycoproteins can adhere to injured
areas of vessel wall.
– The platelet phospholipids can activate multiple
stages in the blood clotting process.
– When platelets come in contact with the exposed
collagen of a damaged endothelium, they release
their granule contents- ADP, TXA2
124.
• Platelet Plug:
–The enables them to adhere to collagen in the
tissues and to a protein called von Willebrand
factor that leaks into the traumatized tissue from
plasma.
– The granule contents also act on nearby platelets
to activate them as well.
– This leads to more activated platelets and the
formation of a platelet plug.
125.
• Platelet Plug:
–The platelet plug is important for closing minute
ruptures in small blood vessels
– Defective platelets will result in micro
haemorrhages like
• Petechiae
• Purpura
• Ecchymosis
126.
• Blood Coagulation:
•Formation of a blood clot starts in 15-20
seconds for severe trauma and 1-2 minutes for
minor trauma.
• Normally, there is a thin balance between the
procoagulants and anticoagulants so that clots
do not form in the blood vessel.
127.
• Blood clottingis initiated by the following
mechanisms:
– Trauma to the vessel wall
– Trauma to adjacent tissues
– Trauma to the blood
– Contact of blood with damaged endothelium or
with collagen
128.
• This leadsto the formation of a prothrombin
activator, which then causes prothrombin
conversion to thrombin.
• The clotting factors are plasma proteins (inactive
proteolytic enzymes)
• Prothrombin activator is formed via:
– Extrinsic pathway- due to trauma to the vessel wall and
surrounding tissues
– Intrinsic pathway: Begins in the blood when collagen is
exposed.
129.
• Extrinsic pathway:
–Tissue trauma leads to release of tissue factor or
tissue thromboplastin (factor III)
– Tissue factor combines with factor VII to activate
factor X (requires calcium ions)
– Xa+ V+III activates prothrombin which is split to
form thrombin.
– Thrombin further activates factor V. Va then
causes further prothrombin activation
130.
• Intrinsic Pathway
•Activation of factor XII and release of platelet
phospholipids.
• XIIa acts on factor XI to activate it. This requires HMW
kinogen or prekallikrein.
• XIa then activates factor IX
• Ixa+VIII+ platelet phospholipids and factor III from
traumatized platelets activates factor X
• X+V+platelet or tissue phospholipids forms the
prothrombin activator.
131.
• Final commonpathway
– Prothrombin is cleaved to thrombin.
– Thrombin acts on fibrinogen to form fibrin
monomers
– Fibrin-stabilizing factor (factor XIII) stabilizes clot
• Clot stabilization: factor XIII cause covalent
bonds between fibrin monomer molecules and
cross-linkages between adjacent fibrin fibers
132.
• Clot retraction:
–Blood clot is composed of fibrin meshwork and
entrapped blood cells, platelets and plasma.
– The fibrin fibers also adhere to damaged surfaces
of blood vessels making the clot to be adherent
and so prevent further blood loss.
– Clot begins to contract few minutes after it was
formed. It discharges most of the fluid from the
clot within 20-60 minutes. This fluid is serum
133.
• Clot retraction
–As clot retracts, the edges of the blood vessel are
pulled together, thus enhancing haemostasis.
– Platelets are necessary for clot retraction.
• They become attach to fibrin fibers and bond different
fibers together.
• They also enhance clot retraction by their contractile
proteins.
• Calcium ions from platelets and thrombin also combine
to enhance clot retraction.
134.
• Positive feedbackof clot formation
– Clot formation initiates a positive feedback to
promote more blood clotting.
– This is enhanced by the direct proteolytic action of
thrombin.
– Thrombin acts on prothrombin converting it to more
thrombin. It also activates the action factor VII,IX,X,XI
and XII as well as platelet aggregation.
– This mechanism causes the blood clot to grow until
further leakage ceases.
135.
• Role ofcalcium
– Apart from the first two steps in the intrinsic
pathway, calcium ions are required for acceleration
of all the blood clotting reactions by either pathway.
– However, in the living body, the calcium ion
concentration seldom falls low enough to
significantly affect the kinetics of blood clotting.
– In vitro, calcium ions in blood can be reduced by
citrate and oxalate ions.
136.
• Role ofvitamin K
– Vitamin K is essential for carboxylation of
prothrombin, factor VII,IX,X and protein C.
– Vitamin K is maintained in its active form by an
enzyme called the reductase enzyme.
– Deficiency of vitamin K occur secondary to poor fat
absorption due to chronic liver disease or
cholestasis.
– A lack of vitamin K can decrease prothrombin
formation resulting in bleeding.
• Intravascular anticoagulants
–Intact endothelium: glycocalyx layer repels clotting
factors and platelets
– Thrombomodulin: binds thrombin and inactivates it.
– Fibrin: adsorbs and removes thrombin from the
blood.
– Antithrombin III- inactivates thrombin
– Heparin: binds AT III and increases the ability of AT
III to remove thrombin.
139.
• Fibrinolysis (PlasminSystem)
– Plasminogen in plasma become activated to plasmin
(fibrinolysin) in the plasma by tissue plasminogen
activator once bleeding is stopped by the clot.
– Plasmin digests fibrin fibers and other clotting
factors such as fibrinogen, factor V,VIII, prothrombin
and factor XII.
– This leads to clot lysis and sometimes
hypercoagulability.
140.
• Bleeding disorders
–Liver failure
– Vitamin K deficiency
– Haemophilia
• Occurs primarily in males
• Due to abnormal or deficient factor VIII (Classic
haemophilia)
• May also be caused by deficiency of factor IX
• Prolonged bleeding, bleeding into muscles and joints.
141.
• Bleeding disorders
–Von Willebrand’s disease: due to deficiency of the
larger fragment of factor VIII.
– Thrombocytopaenia: low platelet count leads to
punctate haemorrhages. Bleeding is usually seen
at counts less than 50,000/cmm.
142.
• Thromboembolism
– Thromboembolicconditions are due to roughened
endothelium and sluggish blood flow.
• Common sites are femoral veins and pulmonary
vessels.
• Disseminated intravascular coagulation:
– due to widespread activation of the clotting
mechanism resulting in bleeding.
– Caused by infection or massive tissue damage
143.
• Anticoagulants
– Heparin
–Coumarins. Eg. Warfarin
– Citrate: binds calcium in the blood
– Oxalate: precipitate calcium ions from blood
144.
• Blood coagulationtests
– Bleeding time: usually prolonged in
thrombocytopaenia. Normal 1-6 minutes
– Clotting time. Normal clotting time is 6-10
minutes.
– Prothrombin time/ International normalized ratio












































































![• In the thymus, the T lymphocytes acquire
specific reactivity against one antigen and the
abilty to recognise self [self tolerance].
• The preprocessing of T lymphocytes in the
thymus occurs shortly before birth and a few
months after birth.
• After preprocessing, the B lymphocytes migrate
to lymphoid tisue throughout the body where
they lodge near the T lymphocyte areas.](https://image.slidesharecdn.com/bloodphysiologylecturenotes1-250408163714-9015e6e6/85/BLOOD-PHYSIOLOGY-LECTURE-NOTES2-1-pptx-79-320.jpg)













































































































![5 Penicillins and Cephalosporines[1].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/5penicillinsandcephalosporines1-250504080824-5bb78aec-thumbnail.jpg?width=640&height=640&fit=bounds)




![ONFH[AVN HIP] -TRIPLE REGIME -A NOVAL SURGICAL CONCEPT .pptx](https://cdn.slidesharecdn.com/ss_thumbnails/onfhavnhip2026koaconcalicutdrgokuldevdrmashraf-260210064517-213ec005-thumbnail.jpg?width=640&height=640&fit=bounds)














