This document provides information about the ninth edition of Apley's System of Orthopaedics and Fractures. It lists the principal authors and contributors, and provides a brief preface and acknowledgements. The book is dedicated to the authors' students, trainees, patients, wives, children and grandchildren. It was first published in 1959 and this ninth edition was published in 2010 by Hodder Arnold.





![iv
First published in Great Britain in 1959 by Butterworths Medical Publications
Second edition 1963
Third edition 1968
Fourth edition 1973
Fifth edition 1977
Sixth edition 1982
Seventh edition published in 1993 by Butterworth Heineman.
Eight edition published in 2001 by Arnold.
This ninth edition published in 2010 by
Hodder Arnold, an imprint of Hodder Education, an Hachette UK Company,
338 Euston Road, London NW1 3BH
http://www.hodderarnold.com
© 2010 Solomon, Warwick, Nayagam
All rights reserved. Apart from any use permitted under UK copyright law, this publica-
tion may only be reproduced, stored or transmitted, in any form, or by any means, with
prior permission in writing of the publishers or in the case of reprographic production, in
accordance with the terms of licences issued by the Copyright Licensing Agency. In the
United Kingdom such licences are issued by the Copyright Licensing Agency:
90 Tottenham Court Road, London W1T 4LP
Whilst the advice and information in this book are believed to be true and accurate at the
date of going to press, neither the author[s] nor the publisher can accept any legal
responsibility or liability for any errors or omissions that may be made. In particular (but
without limiting the generality of the preceding disclaimer) every effort has been made to
check drug dosages; however it is still possible that errors have been missed. Furthermore,
dosage schedules are constantly being revised and new side-effects recognized. For these
reasons the reader is strongly urged to consult the drug companies’ printed instructions
before administering any of the drugs recommended in this book.
British Library Cataloguing in Publication Data
A catalogue record for this book is available from the British Library
Library of Congress Cataloging-in-Publication Data
A catalog record for this book is available from the Library of Congress
ISBN-13 978 0 340 942 055
ISBN-13 [ISE] 978 0 340 942 086 (International Students’ Edition, restricted
territorial availability)
1 2 3 4 5 6 7 8 9 10
Commissioning Editor: Gavin Jamieson
Project Editor: Francesca Naish
Production Controller: Joanna Walker
Cover Designer: Helen Townson
Indexer: Laurence Errington
Additional editorial services provided by
Naughton Project Management.
Cover image © Linda Bucklin/stockphoto.com
Typeset in 10 on 12pt Galliard by Phoenix Photosetting, Chatham, Kent
Printed and bound in India by Replika Press
What do you think about this book? Or any other Hodder Arnold title?
Please visit our website: www.hodderarnold.com](https://image.slidesharecdn.com/apleys-150117201150-conversion-gate02/85/Apley-s-shoulderjt-examination-5-320.jpg)


















![Find out details about the patient’s work practices,
travel and recreation: could the disorder be due to a
particular repetitive activity in the home, at work or
on the sportsfield? Is the patient subject to any
unusual occupational strain? Has he or she travelled to
another country where tuberculosis is common?
Finally, it is important to assess the patient’s home
circumstances and the level of support by family and
friends. This will help to answer the question: ‘What
has the patient lost and what is he or she hoping to
regain?’
EXAMINATION
In A Case of Identity Sherlock Holmes has the follow-
ing conversation with Dr Watson.
Watson: You appeared to read a good deal upon
[your client] which was quite invisible to me.
Holmes: Not invisible but unnoticed, Watson.
Some disorders can be diagnosed at a glance: who
would mistake the facial appearance of acromegaly or
the hand deformities of rheumatoid arthritis for any-
thing else? Nevertheless, even in these cases systematic
examination is rewarding: it provides information
about the patient’s particular disability, as distinct
from the clinicopathological diagnosis; it keeps rein-
forcing good habits; and, never to be forgotten, it lets
the patient know that he or she has been thoroughly
attended to.
The examination actually begins from the moment
we set eyes on the patient. We observe his or her gen-
eral appearance, posture and gait. Can you spot any
distinctive feature: Knock-knees? Spinal curvature? A
short limb? A paralysed arm? Does he or she appear to
be in pain? Do their movements look natural? Do they
walk with a limp, or use a stick? A tell-tale gait may
suggest a painful hip, an unstable knee or a foot-drop.
The clues are endless and the game is played by every-
one (qualified or lay) at each new encounter through-
out life. In the clinical setting the assessment needs to
be more focussed.
When we proceed to the structured examination,
the patient must be suitably undressed; no mere
rolling up of a trouser leg is sufficient. If one limb is
affected, both must be exposed so that they can be
compared.
We examine the good limb (for comparison), then
the bad. There is a great temptation to rush in with
both hands – a temptation that must be resisted. Only
by proceeding in a purposeful, orderly way can we
avoid missing important signs.
Alan Apley, who developed and taught the system
used here, shied away from using long words where
short ones would do as well. (He also used to say ‘I’m
neither an inspector nor a manipulator, and I am defi-
nitely not a palpator’.) Thus the traditional clinical
routine, inspection, palpation, manipulation, was
replaced by look, feel, move. With time his teaching has
been extended and we now add test, to include the
special manoeuvres we employ in assessing neurolog-
ical integrity and complex functional attributes.
Look
Abnormalities are not always obvious at first sight. A
systematic, step by step process helps to avoid mis-
takes.
Shape and posture The first things to catch one’s
attention are the shape and posture of the limb or the
body or the entire person who is being examined. Is
the patient unusually thin or obese? Does the overall
posture look normal? Is the spine straight or unusu-
ally curved? Are the shoulders level? Are the limbs
normally positioned? It is important to look for defor-
mity in three planes, and always compare the affected
part with the normal side. In many joint disorders and
in most nerve lesions the limb assumes a characteristic
posture. In spinal disorders the entire torso may be
deformed. Now look more closely for swelling or
wasting – one often enhances the appearance of the
other! Or is there a definite lump?
Skin Careful attention is paid to the colour, quality
and markings of the skin. Look for bruising, wounds
and ulceration. Scars are an informative record of the
past – surgical archaeology, so to speak. Colour
reflects vascular status or pigmentation – for example
the pallor of ischaemia, the blueness of cyanosis, the
redness of inflammation, or the dusky purple of an old
bruise. Abnormal creases, unless due to fibrosis, sug-
gest underlying deformity which is not always obvi-
ous; tight, shiny skin with no creases is typical of
oedema or trophic change.
GENERALORTHOPAEDICS
6
1
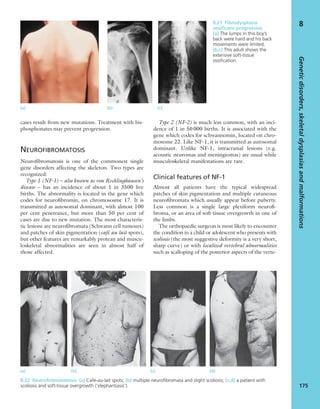
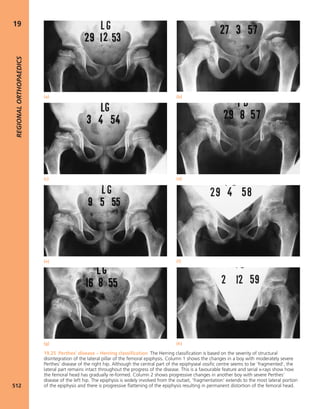
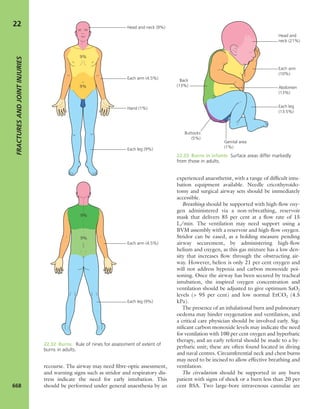
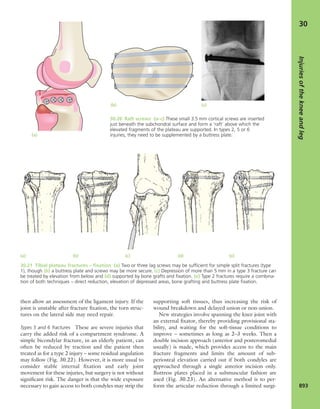
1.3 Look Scars often give clues to the previous history.
The faded scar on this patient’s thigh is an old operation
wound – internal fixation of a femoral fracture. The other
scars are due to postoperative infection; one of the sinuses
is still draining.](https://image.slidesharecdn.com/apleys-150117201150-conversion-gate02/85/Apley-s-shoulderjt-examination-25-320.jpg)


















































































































![periosteum where mesenchymal cells differentiate into
osteoblasts (intramembranous, or ‘appositional’, bone
formation) and old bone is removed from the inside of
the cylinder by osteoclastic endosteal resorption.
Intramembranous periosteal new bone formation
also occurs as a response to periosteal stripping due to
trauma, infection or tumour growth, and its appear-
ance is a useful radiographic pointer.
BONE RESORPTION
Bone resorption is carried out by the osteoclasts under
the influence of stromal cells (including osteoblasts)
and both local and systemic activators. Although it has
long been known that PTH promotes bone resorp-
tion, osteoclasts have no receptor for PTH but the
hormone acts indirectly through its effect on the
vitamin D metabolite 1,25-dihydroxycholecalciferol
[1,25(OH)2D3] and osteoblasts.
Proliferation of osteoclastic progenitor cells
requires the presence of an osteoclast differentiating
factor produced by the stromal osteoblasts after stim-
ulation by (for example) PTH, glucocorticoids or
pro-inflammatory cytokines. It is now known that this
‘osteoclast differentiating factor’ is the receptor acti-
vator of nuclear factor-κβ ligand (RANKL for short),
and that it has to bind with a RANK receptor on the
osteoclast precursor in the presence of a macrophage
colony-stimulating factor (M-CSF) before full matu-
ration and osteoclastic resorption can begin.
It is thought that osteoblasts first ‘prepare’ the
resorption site by removing osteoid from the bone
surface while other matrix constituents act as osteo-
clast attractors. During resorption each osteoclast
forms a sealed attachment to the bone surface where
the cell membrane folds into a characteristic ruffled
border within which hydrochloric acid and proteolytic
enzymes are secreted. At this low pH minerals in the
matrix are dissolved and the organic components are
destroyed by lysosomal enzymes. Calcium and phos-
phate ions are absorbed into the osteoclast vesicles
from where they pass into the extracellular fluid and,
ultimately, the blood stream.
In cancellous bone this process results in thinning
(and sometimes actual perforation) of existing trabec-
ulae. In cortical bone the cells either enlarge an exist-
ing haversian canal or else burrow into the compact
bone to create a cutting cone – like miners sinking a
new shaft in the ground. During hyperactive bone
resorption these processes are reflected in the appear-
ance of hydroxyproline in the urine and a rise in
serum calcium and phosphate levels.
BONE MODELLING AND REMODELLING
The sequential process of bone resorption and forma-
tion has been likened to sculpting and is, in fact,
known as bone modelling and remodelling. During
growth each bone has continuously to be ‘sculpted’
into the normal shape of that particular part of the
skeleton. How else can a long bone retain its basic
shape as the flared ends are constantly re-formed fur-
ther and further from the midshaft during growth?
The internal architecture of the bone is also subject
to remodelling, not only during growth but through-
out life. This serves several crucial purposes: ‘old
bone’ is continually replaced by ‘new bone’ and in this
way the skeleton is protected from exposure to cumu-
lative loading frequencies and the risk of stress failure;
bone turnover is sensitive to the demands of function
GENERALORTHOPAEDICS
122
7
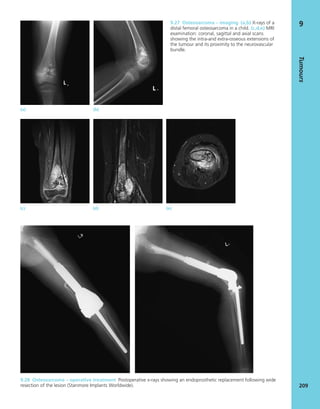
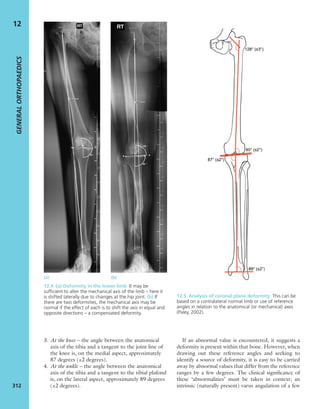
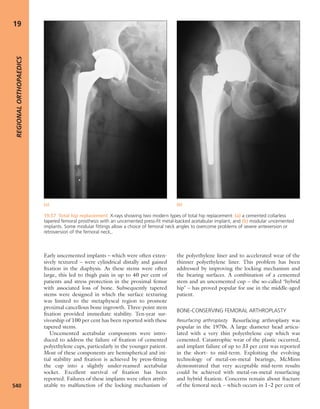
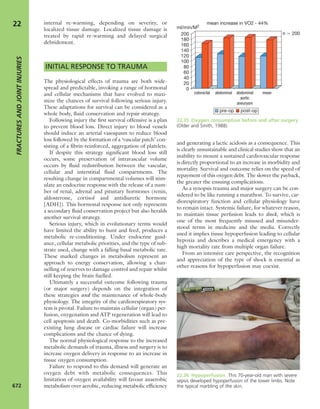
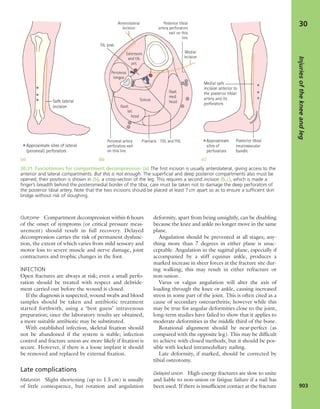
7.4 Blood supply to bone Schematic presentation of
blood supply in tubular bones. (Reproduced from Bullough
PG. Atlas of Orthopaedic Pathology: With Clinical and
Radiological Correlations (2nd edition). Baltimore:
University Park Press, 1985. By kind permission of Dr Peter
G Bullough and Elsevier.)](https://image.slidesharecdn.com/apleys-150117201150-conversion-gate02/85/Apley-s-shoulderjt-examination-141-320.jpg)


![between 3 and 3.5 mmol/L, patients may complain of
anorexia, nausea, muscle weakness and fatigue. Those
with severe hypercalcaemia (more than 3.5 mmol/L)
have a plethora of symptoms including abdominal pain,
nausea, vomiting, severe fatigue and depression. In
longstanding cases patients may develop kidney stones
or nephrocalcinosis due to chronic hypercalciuria;
some complain of joint symptoms, due to
chondrocalcinosis. The clinical picture is aptly (though
unkindly) summarized in the old adage ‘moans,
groans, bones and stones’.
There may also be symptoms and signs of the
underlying cause, which should always be sought (in
the vast majority this will be hyperparathyroidism,
metastatic bone disease, myelomatosis, Paget’s disease
or renal failure).
Phosphorus
Apart from its role (with calcium) in the composition
of hydroxyapatite crystals in bone, phosphorus is
needed for many important metabolic processes,
including energy transport and intracellular cell sig-
nalling. It is abundantly available in the diet and is
absorbed in the small intestine, more or less in pro-
portion to the amount ingested; however, absorption
is reduced in the presence of antacids such as alu-
minium hydroxide, which binds phosphorus in the
gut. Phosphate excretion is extremely efficient, but 90
per cent is reabsorbed in the proximal tubules. Plasma
concentration – almost entirely in the form of ionized
inorganic phosphate (Pi) – is normally maintained at
0.9–1.3 mmol/L (2.8–4.0 mg/dL).
The solubility product of calcium and phosphate is
held at a fairly constant level; any increase in the one
will cause the other to fall. The main regulators of
plasma phosphate concentration are PTH and 1,25-
(OH)2D. If the Pi rises abnormally, a reciprocal fall in
calcium concentration will stimulate PTH secretion
which in turn will suppress urinary tubular reabsorp-
tion of Pi, resulting in increased Pi excretion and a fall
in plasma Pi. High Pi levels also result in diminished
1,25-(OH)2D production, causing reduced intestinal
absorption of phosphorus.
In recent years interest has centred on another group
of hormones or growth factors which also have the ef-
fect of suppressing tubular reabsorption of phosphate
independently of PTH. These so-called ‘phospho-
tonins’ are associated with rare phosphate-losing dis-
orders and tumour-induced osteomalacia. Their exact
role in normal physiology is still under investigation.
Magnesium
Magnesium plays a small but important part in min-
eral homeostasis. The cations are distributed in the
cellular and extracellular compartments of the body
and appear in high concentration in bone. Magne-
sium is necessary for the efficient secretion and
peripheral action of parathyroid hormone. Thus, if
hypocalcaemia is accompanied by hypomagnesaemia
it cannot be fully corrected until normal magnesium
concentration is restored.
Vitamin D
Vitamin D, through its active metabolites, is princi-
pally concerned with calcium absorption and transport
and (acting together with PTH) bone remodelling.
Target organs are the small intestine and bone.
Naturally occurring vitamin D (cholecalciferol) is
derived from two sources: directly from the diet and
indirectly by the action of ultraviolet light on the pre-
cursor 7-dehydrocholesterol in the skin. For people
who do not receive adequate exposure to bright sun-
light, the recommended daily requirement for adults is
400–800 IU (10–20 μg) per day – the higher dose for
people over 70 years of age. In most countries this is
obtained mainly from exposure to sunlight; those who
lack such exposure are likely to suffer from vitamin D
deficiency unless they take dietary supplements.
Vitamin D itself is inactive. Conversion to active
metabolites (which function as hormones) takes place
first in the liver by 25-hydroxylation to form 25-
hydroxycholecalciferol [25-OHD], and then in the
kidneys by further hydroxylation to 1,25-dihydroxy-
cholecalciferol [1,25-(OH)2D]. The enzyme responsi-
ble for this conversion is activated mainly by PTH, but
also by other hormones (including oestrogen and pro-
lactin) or by an abnormally low concentration of phos-
phate. If the PTH concentration falls and phosphate
remains high, 25-OHD is converted alternatively to
24,25-(OH)2D which is inactive. On the other hand,
during negative calcium balance production switches
to 1,25-(OH)2D in response to PTH secretion (see
below); the increased 1,25-(OH)2D then helps to
restore the serum calcium concentration.
Metabolicandendocrinedisorders
125
7
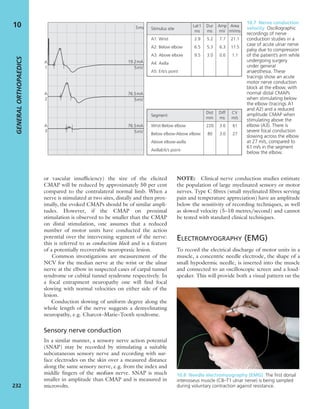
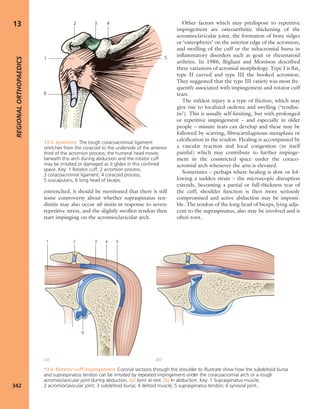
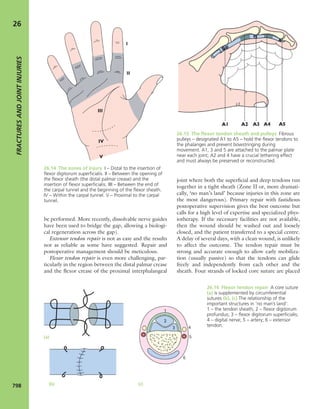
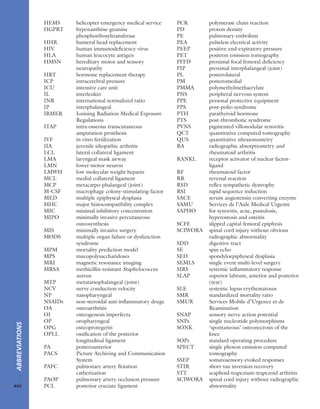
7.7 Vitamin D metabolism The active vitamin D
metabolites are derived either from the diet or by
conversion of precursors when the skin is exposed to
sunlight. The inactive ‘vitamin’ is hydroxylated, first in the
liver and then in the kidney, to form the active metabolites
25-HCC and 1,25-DHCC.](https://image.slidesharecdn.com/apleys-150117201150-conversion-gate02/85/Apley-s-shoulderjt-examination-144-320.jpg)















![fatigue and muscle weakness. Patients may develop
polyuria, kidney stones or nephrocalcinosis due to
chronic hypercalciuria. Some complain of joint symp-
toms, due to chondrocalcinosis. Only a minority
(probably less than 10 per cent) present with bone
disease; this is usually generalized osteoporosis rather
than the classic features of osteitis fibrosa, bone cysts
and pathological fractures.
X-rays
Typical x-ray features are osteoporosis (sometimes
including vertebral collapse) and areas of cortical ero-
sion. Hyperparathyroid ‘brown tumours’ should be
considered in the differential diagnosis of atypical
cyst-like lesions of long bones. The classical – and
almost pathognomonic – feature, which should always
be sought, is sub-periosteal cortical resorption of the
middle phalanges. Non-specific features of hypercal-
caemia are renal calculi, nephrocalcinosis and chon-
drocalcinosis.
Biochemical tests
There may be hypercalcaemia, hypophosphataemia
and a raised serum PTH concentration. Serum alka-
line phosphatase is raised with osteitis fibrosa.
Diagnosis
It is necessary to exclude other causes of hypercal-
caemia (multiple myeloma, metastatic disease, sar-
coidosis) in which PTH levels are usually depressed.
Hyperparathyroidism also comes into the differential
diagnosis of all types of osteoporosis and osteomalacia.
Treatment
Treatment is usually conservative and includes ade-
quate hydration and decreased calcium intake. The
indications for parathyroidectomy are marked and
unremitting hypercalcaemia, recurrent renal calculi,
progressive nephrocalcinosis and severe osteoporosis.
Postoperatively there is a danger of severe hypocal-
caemia due to brisk formation of new bone (the ‘hun-
gry bone syndrome’). This must be treated promptly,
with one of the fast-acting vitamin D metabolites.
SECONDARY HYPERPARATHYROIDISM
Parathyroid oversecretion is a predictable response to
chronic hypocalcaemia. Secondary hyperparathy-
roidism is seen, therefore, in various types of rickets
and osteomalacia, and accounts for some of the radi-
ological features in these disorders. Treatment is
directed at the primary condition.
RENAL OSTEODYSTROPHY
Patients with chronic renal failure and lowered
glomerular filtration rate are liable to develop diffuse
bone changes which resemble those of other condi-
tions that affect bone formation and mineralization.
Thus the dominant picture may be that of secondary
hyperparathyroidism [due to phosphate retention,
hypocalcaemia and diminished production of 1,25-
(OH)2D], osteoporosis, osteomalacia or – in
advanced cases – a combination of these. In older
Metabolicandendocrinedisorders
141
7
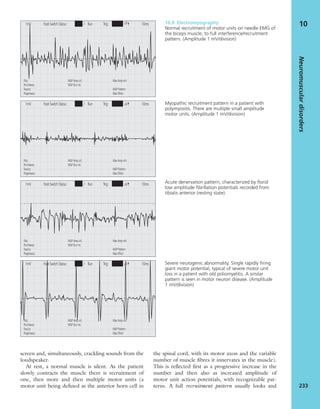
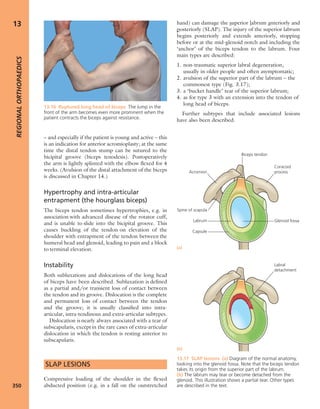
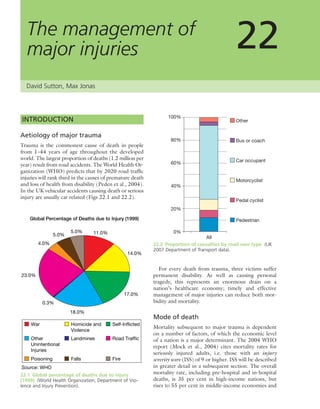
(a) (c)
(b)
7.18 Renal glomerular
osteodystrophy (a) This young
boy with chronic renal failure
developed severe deformities of
(b) the hips and (c) the knees.
Note the displacement of the
upper femoral epiphyses.](https://image.slidesharecdn.com/apleys-150117201150-conversion-gate02/85/Apley-s-shoulderjt-examination-160-320.jpg)