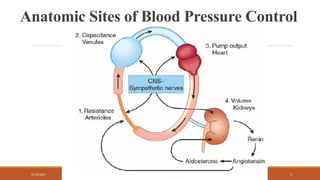
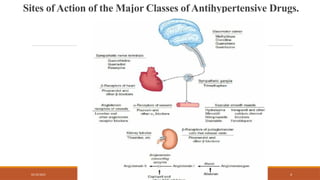
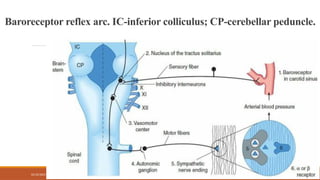
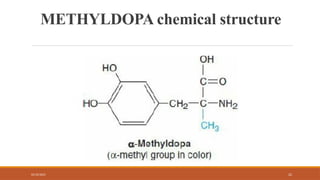
The document provides an overview of antihypertensive agents, detailing their mechanisms of action, classification, and the significance of dietary sodium restriction. It discusses various drug classes including diuretics, sympathoplegic agents, and direct vasodilators, as well as the associated toxicities and effectiveness in treating hypertension. Additionally, it covers the actions and pharmacokinetics of specific drugs like methyldopa and clonidine, along with their side effects and considerations for use.




















































































































![CTEV [ clubfoot] DR ARUN LAL ,DR MOHAMED ASHRAF travancore medical college k...](https://cdn.slidesharecdn.com/ss_thumbnails/ctevclubfootdrarunlaldrmohamedashraftravancoremedicalcollegekollamkeralaindia-260208063247-18fc466c-thumbnail.jpg?width=640&height=640&fit=bounds)









